
Actinide
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | |
| 1 | H | He | ||||||||||||||||
| 2 | Li | Be | B | C | N | O | F | Ne | ||||||||||
| 3 | Na | Mg | Al | Si | P | S | Cl | Ar | ||||||||||
| 4 | K | Ca | Sc | Ti | V | Cr | Mn | Fe | Co | Ni | Cu | Zn | Ga | Ge | As | Se | Br | Kr |
| 5 | Rb | Sr | Y | Zr | Nb | Mo | Tc | Ru | Rh | Pd | Ag | Cd | In | Sn | Sb | Te | I | Xe |
| 6 | Cs | Ba | * | Hf | Ta | W | Re | Os | Ir | Pt | Au | Hg | Tl | Pb | Bi | Po | At | Rn |
| 7 | Fr | Ra | ** | Rf | Db | Sg | Bh | Hs | Mt | Ds | Rg | Cn | Nh | Fl | Mc | Lv | Ts | Og |
| 8 | Uue | Ubn | ⁂ | |||||||||||||||
| ↓ | ||||||||||||||||||
| * | La | Ce | Pr | Nd | Pm | Sm | Eu | Gd | Tb | Dy | Ho | Er | Tm | Yb | Lu | |||
| ** | Ac | Th | Pa | U | Np | Pu | Am | Cm | Bk | Cf | Es | Fm | Md | No | Lr | |||
| ⁂ | Ubu | Ubb | Ubt | Ubq | Ubp | Ubh | Ubs | Ubo | Ube | Utn | Utu | Utb | Utt | Utq | Utp | |||
| Uth | Uts | Uto | Ute | Uqn | Uqu | Uqb | ||||||||||||
| Uue | Éléments hypothétiques | Mt | Nature chimique inconnue | |||||||||||||||
| Li | Métaux alcalins | Al | Métaux pauvres | |||||||||||||||
| Be | Métaux alcalino-terreux | B | Métalloïdes | |||||||||||||||
| Sc | Métaux de transition | Non-métaux : | ||||||||||||||||
| La | Lanthanides (*) | H | « CHNOPS » et sélénium | |||||||||||||||
| Ac | Actinides (**) | F | Halogènes | |||||||||||||||
| Ubu | Superactinides (⁂) | He | Gaz nobles | |||||||||||||||
Les actinides sont une famille du tableau périodique comprenant les quinze éléments chimiques allant de l'actinium (no 89) au lawrencium (no 103). Ces métaux lourds tirent leur nom de l'actinium, premier de la famille, en raison de leurs propriétés chimiques apparentées. On les désigne parfois sous le symbole chimique collectif An, qui représente alors n'importe quel actinide. Ce sont tous des éléments du bloc f, hormis le lawrencium, qui appartient au bloc d. Contrairement aux lanthanides, qui appartiennent également au bloc f, les actinides présentent un nombre de valence sensiblement plus variable. Ils ont tous un rayon atomique et un rayon ionique élevé, et leurs propriétés physiques sont particulièrement diverses. Ainsi, alors que les actinides de numéro atomique élevé se comportent chimiquement comme des lanthanides, ceux du début de la famille, allant du thorium au neptunium, ont une chimie évoquant par certains aspects celle des métaux de transition.
Tous les actinides sont radioactifs, et libèrent de l'énergie par désintégration radioactive. Ils sont tous fissibles en neutrons rapides, et quelques uns en neutrons thermiques. L'uranium, le thorium et le plutonium sont les actinides les plus abondants sur Terre, les deux premiers étant des éléments primordiaux tandis que le troisième est synthétisé par l'industrie nucléaire ; ces trois éléments sont utilisés dans les réacteurs nucléaires ainsi que dans la production d'armes nucléaires. L'américium est le seul élément synthétique à posséder un usage commercial civil, dans les chambres d'ionisation des détecteurs de fumée. Parmi les actinides, seuls le thorium et l'uranium sont présents en quantité significative dans le milieu naturel du fait de la très longue demi-vie de leurs isotopes les plus stables. La désintégration du thorium 232 et de l'uranium 235 produit de l'actinium et du protactinium, qui sont eux-mêmes radioactifs et ne sont donc présents dans la nature que temporairement avant de se désintégrer à leur tour. De faibles quantités de neptunium et peut-être également de plutonium se forment également par transmutation dans les minerais d'uranium. Tous les autres actinides sont exclusivement synthétiques ; on peut cependant trouver des traces de certains d'entre eux dans l'environnement à la suite d'essais nucléaires atmosphériques, comme l'américium, le curium, le berkélium, le californium, l'einsteinium et le fermium[1]. Ils sont produits à partir d'éléments plus légers par capture neutronique.
L'actinide synthétique le plus abondamment produit est le plutonium, notamment le plutonium 239. Cet isotope n'est pas considéré comme un déchet radioactif car il est lui-même un isotope fissile. Mais les réacteurs nucléaires génèrent, en quantité moindre, d'autres actinides qui sont appelés « mineurs ». La qualification de « mineurs » rend compte du fait que ces éléments sont présents en bien moins grande proportion que les actinides majeurs, uranium et plutonium. Les actinides mineurs constituent avec les produits de fission une partie[2] des déchets HAVL, c’est-à-dire les déchets de l'industrie électronucléaire les plus fortement radioactifs.
- Exemples d'actinides
-

-
 Composants en uranium usinés.
Composants en uranium usinés. -
 Sphère en neptunium.
Sphère en neptunium. -
 Plutonium de qualité militaire.
Plutonium de qualité militaire. -
 Américium vu au microscope.
Américium vu au microscope. -
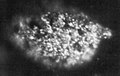 Berkélium (échantillon de 1,7 µg long de 100 µm).
Berkélium (échantillon de 1,7 µg long de 100 µm). -
 Californium (échantillon de 10 mg large d'environ 1 mm).
Californium (échantillon de 10 mg large d'environ 1 mm).

.jpg)





Propriétés[modifier | modifier le code]
Propriétés physiques[modifier | modifier le code]

Les actinides présentent des propriétés semblables à celles des lanthanides. Leurs électrons se répartissent dans les sous-couches 7s et 6d pour les cinq premiers actinides — actinium, thorium, protactinium, uranium et neptunium — et remplissent progressivement la sous-couche 5f à partir du troisième, le protactinium. On observe une diminution progressive du rayon ionique des actinides d'une façon semblable à la contraction des lanthanides.
Les propriétés des actinides sont typiques des métaux. Ce sont tous des matériaux mous aux reflets argentés, mais qui ternissent rapidement à l'air libre. Certains d'entre eux peuvent être coupés au couteau. Ils ont une masse volumique et une plasticité souvent élevées. Leur résistivité varie de 15 à 150 µΩ cm. La dureté du thorium est semblable à celle de l'acier, de sorte que le thorium pur chauffé peut être enroulé en feuillets et étiré en câbles. Le thorium est environ 40 % moins dense que l'uranium et le plutonium mais est plus dur que ces deux éléments. Tous les actinides sont radioactifs, paramagnétiques et, hormis l'actinium, possèdent plusieurs phases cristallines : l'uranium, le neptunium et le californium en ont trois, et le plutonium en a sept. La structure cristalline du protactinium, de l'uranium, du neptunium et du plutonium n'a pas d'équivalent clair parmi les lanthanides et ressemblent davantage à celle des métaux de transition de la 4e période.
Tous les actinides sont pyrophoriques, particulièrement lorsqu'ils sont finement divisés, c'est-à-dire qu'ils s'enflamment spontanément à l'air libre. Leur point de fusion ne dépend pas du nombre de leurs électrons dans la sous-couche 5f ; celui du neptunium et du plutonium, inhabituellement bas à environ 640 °C, s'explique par l'hybridation des orbitales 5f et 6d avec formation de liaisons directionnelles dans ces métaux.
Le tableau ci-dessous résume quelques propriétés physiques des actinides :
Élément Masse
atomiqueTempérature
de fusionTempérature
d'ébullitionMasse
volumiqueRayon de
covalenceConfiguration
électronique[3]Énergie
d'ionisationÉlectronégativité
(Pauling)Actinium [227] 1 227 °C 3 200 ± 300 °C 10 g cm−3 215 pm [Rn] 7s2 6d1 ( * ) 499 kJ mol−1 1,1 Thorium 232,037 7 u 1 750 °C 4 788 °C 11,7 g cm−3 206 ± 6 pm [Rn] 7s2 6d2 ( * ) 587 kJ mol−1 1,3 Protactinium 231,035 88 u 1 568 °C 4 027 °C 15,37 g cm−3 200 pm [Rn] 7s2 5f2 6d1 ( * ) 568 kJ mol−1 1,5 Uranium 238,028 91 u 1 132,2 °C 4 131 °C 19,1 g cm−3 196 ± 7 pm [Rn] 7s2 5f3 6d1 ( * ) 597,6 kJ mol−1 1,38 Neptunium [237] 639 ± 3 °C 4 174 °C 19,38 g cm−3 190 ± 1 pm [Rn] 7s2 5f4 6d1 ( * ) 604,5 kJ mol−1 1,36 Plutonium [244] 639,4 °C 3 228 °C 19,816 g cm−3 187 ± 1 pm [Rn] 7s2 5f6 584,7 kJ mol−1 1,28 Américium [243] 1 176 °C 2 607 °C 12 g cm−3 180 ± 6 pm [Rn] 7s2 5f7 578 kJ mol−1 1,3 Curium [247] 1 340 °C 3 110 °C 13,51 g cm−3 169 ± 3 pm [Rn] 7s2 5f7 6d1 ( * ) 581 kJ mol−1 1,3 Berkélium [247] 986 °C 2 627 °C 13,25 g cm−3 170 pm [Rn] 7s2 5f9 601 kJ mol−1 1,3 Californium [251] 900 °C 1 470 °C 15,1 g cm−3 — [Rn] 7s2 5f10 608 kJ mol−1 1,3 Einsteinium [252] 860 °C 996 °C 8,84 g cm−3 — [Rn] 7s2 5f11 619 kJ mol−1 1,3 Fermium [257] 1 527 °C — 9,7(1) g cm−3 — [Rn] 7s2 5f12 627 kJ mol−1 1,3 Mendélévium [258] 827 °C — 10,3(7) g cm−3 — [Rn] 7s2 5f13 — 1,3 Nobélium [259] 827 °C — 9,9(4) g cm−3 — [Rn] 7s2 5f14 641,6 kJ mol−1 1,3 Lawrencium [266] 1 627 °C — ~15,6–16,6 g cm−3 — [Rn] 7s2 5f14 7p1 ( * ) 478,6 kJ mol−1 —
- ( * ) Exceptions à la règle de Klechkowski : actinium 89Ac, thorium 90Th, protactinium 91Pa, uranium 92U, neptunium 93Np, curium 96Cm et lawrencium 103Lr.
Propriétés chimiques[modifier | modifier le code]
Les actinides réagissent encore plus facilement que les lanthanides avec les halogènes (17e groupe du tableau périodique) et les chalcogènes (16e groupe). Ceux à faible nombre d'électrons dans leur sous-couche 5f sont facilement enclins à l'hybridation. Cela s'explique par les niveaux d'énergie très voisins entre sous-couches 7s, 5f et 6d. La plupart des actinides présentent une grande variété d'états d'oxydation, les plus stables étant l'état +6 pour l'uranium, +5 pour le protactinium et le neptunium, +4 pour le thorium et le plutonium, et +3 pour l'actinium et les autres actinides.
- Actinium — Du point de vue chimique, il est semblable au lanthane, ce qui s'explique par leur rayon ionique et leur configuration électronique semblables. Il possède également l'état d'oxydation +3 mais est moins réactif et présente des propriétés basiques plus prononcées. Ac3+ est plus acide des actinides trivalents, c'est-à-dire que c'est celui qui a le moins tendance à l'hydrolyse en solution aqueuse.
- Thorium — Il est chimiquement plutôt réactif. Ses composés tétravalents sont incolores en raison de l'absence d'électrons dans les sous-couches 5f et 6d. À pH < 3, les solutions de sels de thorium sont dominées par les cations [Th(H2O)8]4+. L'ion Th4+ est relativement gros, avec un rayon variant de 95 pm à 114 pm en fonction du nombre de coordination. Les sels de thorium ont par conséquent une faible tendance à s'hydrolyser. Leur particularité est d'être extrêmement solubles, non seulement dans l'eau mais également dans les solvants organiques polaires.
- Protactinium — Il présente deux états d'oxydation : +5 est stable, tandis que +4 s'oxyde facilement en protactinium(V). Une solution de protactinium tétravalent s'obtient par conséquent par l'action d'un réducteur fort sous atmosphère d'hydrogène. Le protactinium(IV) est chimiquement semblable à l'uranium(IV) et au thorium(IV). Les fluorures, phosphates, hypophosphates et iodates de protactinium(IV) sont insolubles dans l'eau et les acides dilués. Le protactinium forme en revanche des carbonates solubles. Les caractéristiques de l'hydrolyse des composés de protactinium(V) sont semblables à celles des composés de tantale(V) et de niobium(V). Les propriétés chimiques complexes du protactinium résultent du fait que c'est à son niveau que la sous-couche 5f commence à se remplir.
- Uranium — Il présente une valence de 3 à 6, cette dernière étant la plus stable. À l'état hexavalent, l'uranium est très semblable aux éléments du groupe 6. De nombreux composés d'uranium(IV) et d'uranium(VI) sont non stœchiométriques, c'est-à-dire que leur composition chimique n'est pas constante et présente un écart variable à la stœchiométrie. Ainsi, la formule chimique exacte du dioxyde d'uranium est UO2+δ, où δ varie de -0,4 à +0,32. Les composés d'uranium(VI) sont des oxydants faibles. La plupart d'entre eux contiennent le groupe linéaire uranyle UO22+. De quatre à six ligands peuvent se loger dans un plan équatorial perpendiculaire au groupe uranyle. Ce dernier se comporte comme un acide dur et forme des complexes plus forts avec des ligands donneurs d'oxygène qu'avec des ligands donneurs d'azote. Les ions NpO22+ et PuO22+ sont également des formes courantes du neptunium et du plutonium à l'état d'oxydation +6. Les composés d'uranium(VI) présentent des propriétés réductrices, c'est-à-dire qu'ils sont facilement oxydés par l'oxygène de l'air. L'uranium(III) est un réducteur fort. Grâce à la présence d'électrons dans la sous-couche 6d, l'uranium forme des composés organométalliques tels que UIII(C5H5)3 et UIV(C5H5)4.
- Neptunium — Il présente des valences allant de 3 à 7 pouvant être observées simultanément en solution. L'état d'oxydation le plus stable est +5, mais la valence 4 est la plus fréquente dans les composés solides du neptunium. Le neptunium métallique est très réactif. Les ions de neptunium tendent à former des composés de coordination et sont facilement hydrolysés.
- Plutonium — Il présente également des valences allant de 3 à 7 et est de ce fait chimiquement semblable au neptunium et à l'uranium. Il est très réactif et il se forme rapidement un film d'oxyde sur ses surfaces exposées à l'air libre. Il réagit avec l'hydrogène même à des températures aussi basses que 25 à 50 °C. Il forme facilement des halogénures et des alliages intermétalliques. Le plutonium(V) peut prendre part à des réactions de polymérisation. Les réactions d'hydrolyse des ions de plutonium à différents états d'oxydation sont assez variables.
- Américium — Il présente la plus grande diversité chimique parmi les actinides, avec des valences allant de 2 à 6. L'américium divalent s'observe uniquement dans les composés secs et les solutions non aqueuses (acétonitrile CH3CN). Les états d'oxydation +3, +5 et +6 s'observent typiquement en solution aqueuse, mais aussi à l'état solide. L'américium tétravalent forme des composés solides stables (dioxyde, fluorure et hydroxyde) ainsi que des complexes en solution aqueuse. Sa valence la plus stable est 3 en solution aqueuse mais 3 ou 4 à l'état solide.
La valence 3 est la plus stable pour tous les éléments suivant l'américium jusqu'au lawrencium, hormis peut-être pour le nobélium. Le curium peut être tétravalent dans les solides (fluorures, dioxyde). Le berkélium présente, dans les fluorures et le dioxyde solides, une valence 4 plus stable que celle du curium, en plus de la valence 3 ; la stabilité de l'ion Bk4+ en solution est semblable à celle de l'ion Ce4+. La valence 3 est la seule à avoir été observée pour le californium, l'einsteinium et le fermium. La valence 2 a été observée pour le mendélévium et le nobélium ; dans ce dernier cas, elle est plus stable que la valence 3. Le lawrencium présente une valence 3 aussi bien en solution que dans les composés solides.
-
 Nitrate d'uranyle UO2(NO3)2.
Nitrate d'uranyle UO2(NO3)2. -

-

-

-
 Tétrachlorure d'uranium UCl4.
Tétrachlorure d'uranium UCl4. -
 Hexafluorure d'uranium UF6 en ampoule scellée.
Hexafluorure d'uranium UF6 en ampoule scellée. -
 Octaoxyde de triuranium U3O8.
Octaoxyde de triuranium U3O8. -
 Yellowcake dans un tube à essais.
Yellowcake dans un tube à essais.







Radioactivité[modifier | modifier le code]
Isotopes les plus stables des actinides[4] Élément Radioisotope Demi-vie Actinium 227Ac 21,772(3) a Thorium 232Th 14,05(6) Ga Protactinium 231Pa 32,76(11) ka Uranium 238U 4,468(3) Ga Neptunium 237Np 2 144(7) ka Plutonium 244Pu 80(0,9) Ma Américium 243Am 7,37(4) ka Curium 247Cm 15,6(5) Ma Berkélium 247Bk 1,38(25) ka Californium 251Cf 900(40) a Einsteinium 252Es 471,7(1,9) j Fermium 257Fm 100,5(0,2) j Mendélévium 258Md 51,5(0,3) j Nobélium 259No 58(5) min Lawrencium 262Lr 4(1) h
On connaît généralement un grand nombre d'isotopes pour chacun des actinides. Tous ces isotopes sont radioactifs (radioisotopes), et presque tous sont synthétiques. Seuls le thorium 232, l'uranium 235 et l'uranium 238 sont des nucléides primordiaux, tandis que le thorium 230, le protactinium 231 et l'uranium 234 sont présents en quantités significatives dans l'environnement comme produits de désintégration transitoires à longue demi-vie de l'uranium naturel. Ainsi, le thorium naturel est composé de 99,98(2) % de 232Th et de 0,02(2) % 230Th, le protactinium naturel est composé à 100 % de 231Pa, et l'uranium naturel est composé de 0,0054(5) % de 234U, 0,7204(6) % de 235U, et 99,2742(10) % de 238U.
- Thorium — On connaît 30 isotopes du thorium, du 209Th au 238Th. Le thorium 232 est le plus stable, avec une demi-vie de 14,05 milliards d'années, légèrement supérieure à la valeur généralement acceptée pour l'âge de l'univers. Il se désintègre à travers une chaîne de désintégration comptant 12 nucléides et aboutissant au plomb 208. Les autres isotopes sont sensiblement plus instables : le thorium 230 a une demi-vie de 75 380 ans, le thorium 229 a une demi-vie de 7 340 ans, et le thorium 238 a une demi-vie de 1,92 ans. Tous les autres ont une demi-vie de moins de 30 jours, et la plupart ont une demi-vie de moins de 10 min. L'isomère 229mTh est caractérisé par une énergie d'excitation particulièrement faible, de l'ordre de 7,8 ± 0,5 eV.
- Uranium — On connaît 28 isotopes de l'uranium, du 215U au 242U. L'uranium 238 est le plus stable, avec une demi-vie de 4,468 3 milliards d'années, proche de l'âge de la Terre. C'est un émetteur α dont la chaîne de désintégration compte 18 nucléides et aboutit au plomb 206. L'uranium 235 a une chaîne de désintégration de 15 nucléides aboutissant au plomb 207. Ces deux isotopes sont intéressants pour l'industrie nucléaire ainsi que pour la production d'armes nucléaires car 235U est le seul isotope fissile disponible naturellement en quantité appréciable, tandis que 238U est fertile : il se transforme en 239U par capture neutronique, lequel se transmute en neptunium 239 puis en plutonium 239, ce dernier étant fissile.
- Plutonium — On connaît 20 isotopes du plutonium, du 228Pu au 247Pu. Les plus stables sont le plutonium 244, avec une demi-vie de 80,8 millions d'années, le plutonium 242, avec une demi-vie de 373 300 ans, et le plutonium 239, avec une demi-vie de 24 110 ans. Tous les autres isotopes du plutonium ont une demi-vie inférieure à 7 000 ans. Parmi tous ces isotopes, 239Pu et 241Pu sont fissiles, ce dernier se désintégrant également en américium 241 par émission β− avec une période de 14 ans. L'isotope 239Pu est l'un des trois matériaux fissiles utilisés pour produire des armes nucléaires et alimenter des réacteurs nucléaires. Par ailleurs, on connaît également huit isomères, dont aucun n'a une demi-vie supérieure à 1 s.
Composés[modifier | modifier le code]
Oxydes et hydroxydes[modifier | modifier le code]

Certains actinides peuvent exister sous plusieurs formes oxydées telles que An2O3, AnO2, An2O5 et AnO3, où An symbolise un actinide quelconque. Les trioxydes AnO3 sont amphotères pour tous les actinides, tandis que les oxydes An2O3, AnO2 et An2O5 sont basiques, réagissant facilement avec l'eau pour donner des hydroxydes basiques :
- An2O3 + 3 H2O → 2 An(OH)3.
Ces bases sont faiblement solubles dans l'eau et leur activité est proche de celle des hydroxydes des terres rares. La base la plus forte est celle de l'actinium. Tous les composés de l'actinium sont incolores, hormis le sulfure d'actinium Ac2S3. Les dioxydes d'actinides tétravalents cristallisent dans le système cristallin cubique, avec la même structure cristalline que le fluorure de calcium CaF2.
Le thorium réagit avec l'oxygène pour former exclusivement du dioxyde de thorium ThO2 :
Le dioxyde de thorium est un minéral réfractaire dont le point de fusion vaut 3 390 °C, le plus élevé connu pour un oxyde. L'addition de 0,8 à 1 % de ThO2 à du tungstène en stabilise la structure, ce qui permet de renforcer les filaments de tungstène pour les rendre plus résistants aux vibrations. Le ThO2 doit être chauffé de 500 à 600 °C pour le dissoudre dans l'acide, tandis que son chauffage au-dessus de 600 °C produit une forme de dioxyde de thorium très résistante aux acides. L'addition d'une petite quantité d'ions fluorure F− catalyse la dissolution du dioxyde de thorium dans les acides.
On connaît deux oxydes de protactinium : le dioxyde noir PaO2 et l'oxyde blanc Pa2O5 ; le premier est isomorphe avec le dioxyde de thorium ThO2 mais le second est le plus facile à produire. Ces deux oxydes sont basiques, et l'hydroxyde Pa(OH)5 est une base faible faiblement soluble.
La décomposition de certains sels d'uranium à l'air libre à 400 °C, par exemple le nitrate d'uranyle hydraté UO2(NO3)·6H2O, donne le trioxyde d'uranium UO3, de couleur orange. Cet oxyde est amphotère et forme plusieurs hydroxydes, dont le plus stable est UO2(OH)2. La réduction du trioxyde d'uranium par l'hydrogène conduit au dioxyde d'uranium UO2, dont les propriétés sont semblables à celle du dioxyde de thorium ThO2. Cet oxyde est également basique, donnant l'hydroxyde d'uranium U(OH)4.
Le neptunium, le plutonium et l'américium forment deux types d'oxydes basiques : An2O3 et AnO2. Le trioxyde de neptunium NpO3 est instable, de sorte qu'il n'est possible de produire que du Np3O8. Cependant, les oxydes de neptunium et de plutonium de formules génériques AnO2 et An2O3 sont bien caractérisés.
Sels[modifier | modifier le code]

Les actinides réagissent facilement avec les halogènes pour former des sels de formules génériques AnX3 et AnX4, où An représente un actinide quelconque et X un halogène quelconque. Le premier composé de berkélium a été synthétisé en 1962 sous la forme de 3 ng du chlorure BkCl3. Les chlorures, bromures et iodures d'actinides sont solubles dans l'eau tandis que les fluorures sont insolubles, comme c'est également le cas pour les sels correspondants de terres rares. L'uranium forme facilement un hexafluorure incolore, l'hexafluorure d'uranium UF6, qui se sublime à 56,5 °C ; cette propriété le rend utile pour séparer les isotopes de l'uranium par centrifugation en phase gazeuse ou par diffusion gazeuse. Les hexafluorures d'actinides ont des propriétés semblables à celles des anhydrides. Ils sont très sensibles à l'humidité et s'hydrolysent en formant des composés AnO2F2. Le pentachlorure d'uranium UCl5 et l'hexachlorure d'uranium (en) UCl6 ont été synthétisés mais sont tous les deux instables.
Lorsqu'ils réagissent avec des actinides, les acides donnent des sels ; lorsque ces acides ne sont pas oxydants, l'actinide reste dans un état d'oxydation peu élevé :
Cependant, l'hydrogène généré au cours de ces réactions peut réagir avec l'actinide pour former l'hydrure correspondant. L'uranium réagit avec les acides et l'eau bien plus facilement que le thorium.
Les sels d'actinides peuvent également être obtenus par dissolution des hydroxydes correspondants dans des acides. Les nitrates, chlorures, sulfates et perchlorates d'actinides sont solubles dans l'eau. Lorsqu'ils cristallisent à partir d'une solution aqueuse, ces sels forment des hydrates tels que Th(NO3)4·6H2O, Th(SO4)2·9H2O et Pu2(SO4)3·7H2O. Les sels d'actinides à valence élevée s'hydrolysent facilement. Les sulfate, chlorure, perchlorate et nitrate de thorium donnent ainsi des sels basiques tels que Th(OH)2SO4 et Th(OH)3NO3. La solubilité des actinides trivalents et tétravalents suit celle des sels de lanthanides. Les phosphates, fluorures, oxalates, iodates et carbonates d'actinides sont ainsi faiblement solubles dans l'eau ; ils précipitent sous forme d'hydrates, comme ThF4·3H2O et Th(CrO4)2·3H2O.
Les actinides à l'état d'oxydation +6 — hormis les cations de type AnO22+ — forment des anions complexes tels que [AnO4]2− ou encore [An2O7]2−, par exemple. Ainsi, l'uranium, le neptunium et le plutonium forment des sels de type Na2UO4 (uranate) et (NH4)2U2O7 (diuranate). Par rapport aux lanthanides, les actinides donnent plus facilement des composés de coordination, et ce d'autant plus que leur valence est élevée. Les actinides trivalents ne forment pas de fluorures coordonnés, tandis que le thorium tétravalent forme les complexes K2ThF6, KThF5 et même K5ThF9. Le thorium forme également les sulfates (par exemple Na2SO4·Th(SO4)2·5H2O), nitrates et thiocyanates correspondants. Les sels de formule générique An2Th(NO3)6·nH2O sont coordonnés, avec une coordinence égale à 12 pour le thorium. Les actinides pentavalents et hexavalents produisent des sels complexes encore plus facilement. Les composés de coordination d'actinides — thorium et uranium tétravalents — les plus stables s'obtiennent à partir de dicétones telles que l'acétylacétone H3C–CO–CH2–CO–CH3.
Toxicité[modifier | modifier le code]
Les actinides sont des éléments chimiques toxiques, c'est-à-dire que le corps humain exposé à des actinides ou à leurs composés est susceptible de subir des lésions et de développer des maladies. Cette toxicité résulte à la fois des propriétés chimiques et de la radioactivité des actinides, de sorte qu'elle est très variable en nature et en intensité d'un élément à l'autre.
Toxicité chimique[modifier | modifier le code]
- Thorium — Il présente une toxicité chimique limitée en raison de la faible solubilité de ses composés, notamment du dioxyde de thorium ThO2, dans l'eau. Certains de ces composés sont cependant modérément toxiques. La manipulation de tels composés peut entraîner des dermatites. Les symptômes peuvent apparaître des dizaines d'années après avoir été en contact avec ces composés. Par ailleurs, le thorium métallique pulvérulent est pyrophorique et prend feu spontanément à l'air libre.

- Uranium — L'essentiel de l'uranium ingéré par voie orale est excrété à l'issue de la digestion. Environ 0,5 % de sa fraction insoluble (notamment ses oxydes) et 5 % de sa fraction soluble (essentiellement sous forme d'ions uranyle UO22+) peuvent être absorbée au cours du processus. Cependant, ce sont les composés insolubles de l'uranium qui sont les plus dangereux, notamment lorsqu'ils sont inhalés sous formes d'aérosols, car ils se fixent dans les poumons, d'où ils diffusent ensuite dans le reste du corps. Une fois passé dans le sang, l'uranium tend à s'accumuler préférentiellement dans les os en raison de son affinité pour les phosphates ; il y demeure alors pendant plusieurs années. Les effets sur la santé sont multiples et se manifestent par des altérations du fonctionnement des reins, du cerveau, du foie, du cœur et d'autres systèmes physiologiques en raison de la toxicité chimique de l'uranium[5]. L'uranium est également reprotoxique[6],[7]. Des études en laboratoire sur modèle animal ont montré que les ions uranyle UO22+, provenant par exemple de trioxyde d'uranium UO3, de nitrate d'uranyle UO2(NO3)2 ou d'autres composés d'uranium hexavalent, provoquent des maladies congénitales et des atteintes au système immunitaire[8]. Parmi les composés de l'uranium, l'hexafluorure d'uranium UF6, très utilisé dans l'industrie nucléaire pour l'enrichissement de l'uranium, présente un risque particulier en raison de la formation d'acide fluorhydrique au contact des tissus biologiques. Enfin, l'uranium métallique pulvérulent est pyrophorique, ce qui induit un risque d'incendie dans la mesure où des grains de petite taille prennent feu spontanément à l'air libre et à température ambiante.
- Plutonium — Chez l'homme, le plutonium absorbé est transporté par des transferrines et est stocké dans le sang par la ferritine pour finir par s'accumuler dans les poumons, le foie et les os. Un moyen d'en limiter les effets et d'injecter un complexe calcium-DTPA dans les 24 h suivant la contamination, ce qui limite la fixation du plutonium — ainsi que de l'américium et du curium. Étant un élément synthétique produit avant tout pour sa radioactivité, le plutonium est surtout connu pour sa radiotoxicité ; il présente cependant une toxicité chimique semblable à celle des autres métaux lourds.
Radiotoxicité[modifier | modifier le code]

Comme toutes les substances radioactives, les actinides peuvent provoquer des lésions des tissus par contamination superficielle de la peau, par exposition interne résultant de l'ingestion de radioisotopes, et par exposition externe essentiellement aux radiations β et aux rayons γ. Le rayonnement α ne traverse pas la peau, mais peut en revanche traverser les muqueuses des organes internes.
- Actinium — Avec le radium et les transuraniens, c'est l'un des éléments présentant la radioactivité la plus dangereuse, avec une activité spécifique particulièrement élevée pour les radiations α. La principale caractéristique de l'actinium est de s'accumuler dans les couches superficielles des os et d'y demeurer. Lors des phases initiales de l'empoisonnement à l'actinium, cet élément s'accumule également dans le foie. Il est particulièrement dangereux par le fait que le temps nécessaire à son excrétion par le corps humain est supérieur à celui de sa désintégration radioactive, de sorte que la majeure partie de l'actinium absorbé se désintègre dans l'organisme. Son absorption par l'appareil digestif est cependant très inférieure à celle du radium.
- Thorium — Étant un élément naturel, le thorium est présent partout en très faibles quantités, de sorte que le corps humain en contient typiquement de l'ordre de 100 μg et en absorbe 3 μg par jour. 200 ppm du thorium absorbé est fixé par l'organisme, les trois quarts dans les os. Il se désintègre très lentement et émet des rayons α qui sont dangereux s'ils proviennent de l'intérieur du corps. L'inhalation d'aérosols contaminés expose ainsi à un risque accru de cancer du poumon, de cancer du pancréas et de leucémie, tandis que l'ingestion de nourriture contaminée expose à un risque accru de cancer du foie. Si la radiotoxicité du thorium lui-même est assez limitée, le thorium 232, son isotope principal, se désintègre en donnant des nucléides tels que le radium et le radon qui sont sensiblement plus dangereux. C'est la raison pour laquelle l'utilisation de dioxyde de thorium ThO2 dans les manchons à incandescence présente un risque de contamination radioactive car les oxydes des radionucléides 228Ra, 224Ra, 212Pb et 212Bi présentent un point de fusion inférieur à celui du ThO2 et sont donc vaporisés lors de l'utilisation du manchon, conduisant à l'inhalation de radium.
- Protactinium — Il tend à s'accumuler dans les reins et les os. La dose maximum toléré par le corps humain est de 0,03 µCi, ce qui correspond environ à 0,5 µg de 231Pa.
- Uranium — La radiotoxicité de l'uranium résulte de ses émissions α. Elle suit sa toxicité chimique (voir plus haut) et devient prépondérante pour des enrichissements en uranium 235 supérieurs à 6 % ; la toxicité chimique de l'uranium est prépondérante en dessous de ce seuil d'enrichissement, comme c'est notamment le cas pour l'uranium appauvri.
- Plutonium — Il s'accumule dans les poumons, le foie et les os quel que soit le mode de contamination — par voie aérienne, par voie alimentaire ou à la suite d'une blessure avec un objet contaminé — et y demeure pour des décennies. À peine un dixième de la quantité absorbée atteint d'autres organes. La persistance du plutonium dans l'organisme s'explique en partie par sa faible solubilité dans l'eau. Certains isotopes du plutonium émettent des rayonnements α qui provoquent des lésions dans les cellules environnantes. Plusieurs méthodes, donnant des résultats très différents, permettent d'approcher la dose maximum tolérée par l'organisme avant de développer des maladies. Ainsi, la dose létale médiane du plutonium après 30 jours par injection intraveineuse chez le chien est de 0,32 mg kg−1, ce qui permet d'estimer la dose létale pour un homme de 70 kg à environ 22 mg de plutonium. La quantité correspondante pour une contamination par voie aérienne devrait être environ quatre fois cette dose. À l'inverse, la dose maximum pouvant être tolérée sans effet toxique peut être estimée en évaluant la toxicité du plutonium à 2 % de celle du radium, ce qui conduit à une dose maximum tolérable de 5 µg, soit 0,3 µCi. Une quantité si faible est à peine visible au microscope. Les études sur modèle animal ont conduit à réduire encore davantage cette valeur, à seulement 0,65 µg, soit 0,04 µCi. Ces études ont montré que la voie de contamination la plus dangereuse est la voie aérienne, à la suite de laquelle entre 5 et 25 % de la quantité inhalée se fixe dans l'organisme. En fonction de la taille des particules et de la solubilité des composés du plutonium, la contamination se fixe dans les poumons, dans le système lymphatique, ou passe dans la circulation sanguine jusqu'à atteindre le foie et les os. La contamination alimentaire est la voie la moins probable. Dans ce cas, environ 0,05 % des composés de plutonium solubles et 0,01 % des composés insolubles passent dans le sang, tandis que le reste est excrété. L'exposition d'une lésion cutanée à du plutonium conduit en revanche à absorber la presque totalité de celui-ci.
Abondance naturelle et minerais[modifier | modifier le code]


Le thorium et l'uranium sont les deux actinides les plus abondants dans le milieu naturel, avec une fraction pondérale respective de 1,6 × 10-5 et 4 × 10-6[9]. L'uranium est présent dans l'écorce terrestre sous la forme d'un mélange d'oxydes entrant dans la composition de la pechblende, ou uraninite. Il existe des dizaines d'autres minéraux contenant de l'uranium, comme la carnotite K2(UO2)2(VO4)2·3H2O et l'autunite Ca(UO2)2(PO4)2·10-12H2O. La composition isotopique de l'uranium est de 99,274 % d'uranium 238, 0,7204 % d'uranium 235 et 0,0054 % d'uranium 234 ; l'isotope 238U possède la demi-vie la plus longue : 4,51 milliards d'années. La production mondiale d'uranium en 2015 s'élevait à 60 496 t, dont 23 800 t au Kazakhstan et 13 325 t au Canada, tandis que les réserves mondiales en 2013 s'élevaient à 5 902 900 t, dont 29 % en Australie et 12 % au Kazakhstan[10].
Les minéraux les plus abondants contenant du thorium sont la thorianite ThO2, la thorite (Th,U)SiO4 et la monazite (Ce,La,Nd,Th)PO4. La plupart des minéraux à thorium contiennent également de l'uranium, tandis que la plupart des minéraux à uranium contiennent également du thorium ; ces minéraux contiennent également une fraction de lanthanides.
L'abondance pondérale de l'actinium dans l'écorce terrestre n'est que de 5 × 10-17. On le trouve dans les minéraux contenant de l'uranium, le plus souvent dans une proportion correspondant à l'équilibre isotopique avec l'uranium 235, son isotope parent. Le protactinium est plus abondant que l'actinium, avec une abondance pondérale d'environ 10-14. Sa concentration dans les minerais d'uranium suit celle de l'uranium 235.
La demi-vie du neptunium 237, isotope le plus stable du neptunium, est négligeable comparée à l'âge de la Terre, de sorte que cet élément n'existe dans le milieu naturel que comme produit intermédiaire de désintégration d'autres isotopes radioactifs. L'abondance naturelle du plutonium 240, isotope le plus stable du plutonium, est de 3 × 10-22 : son extrême rareté fait que le plutonium utilisé dans l'industrie nucléaire et dans celle de l'armement est entièrement synthétisé artificiellement.
Extraction[modifier | modifier le code]
En raison de la faible abondance naturelle des actinides, leur extraction à partir des minerais passe par des procédés complexes en plusieurs étapes. On utilise généralement des fluorures comme produits intermédiaires car ils sont insolubles dans l'eau et peuvent être facilement purifiés par des réactions d'oxydoréduction. Les fluorures sont réduits par le calcium, le magnésium et le baryum :
- 2 AmF3 (en) + 3 Ba → 3 BaF2 + 2 Am, de ;
- PuF4 + 2 Ba → 2 BaF2 + Pu, 1 200 °C ;
- UF4 + 2 Mg → U + 2 MgF2, > 500 °C.
La principale difficulté de l'extraction de l'actinium, par exemple, est la grande similitude de ses propriétés avec celles du lanthane, ce qui fait qu'il est généralement synthétisé par des réactions nucléaires à partir d'isotopes du radium, ou alors séparé à l'aide de procédés par échange d'ions.
Extraction du thorium[modifier | modifier le code]
Le thorium et l'uranium sont les premiers actinides à pouvoir être isolés. Le thorium est essentiellement extrait de la monazite. On fait réagir le pyrophosphate de thorium ThP2O7 avec de l'acide nitrique HNO3, ce qui donne du nitrate de thorium Th(NO3)4, lequel est traité au phosphate de tributyle (CH3CH2CH2CH2O)3PO. Les terres rares également présentes dans la monazite sont éliminées en augmentant le pH dans une solution de sulfate.
Dans une autre méthode d'extraction, la monazite est décomposée avec une solution aqueuse d'hydroxyde de sodium NaOH à 140 °C. Les hydroxydes métalliques sont extraits en premier, filtrés à 80 °C, lavés à l'eau et dissous dans l'acide chlorhydrique concentré. La solution acide est alors neutralisée avec des hydroxydes jusqu'à pH = 5,8, ce qui conduit à la formation d'un précipité d'hydroxyde de thorium Th(OH)4 contenant environ 3 % d'hydroxydes de terres rares, le reste des hydroxydes de terres rares demeurant en solution. L'hydroxyde de thorium est dissous dans un acide minéral et purifié de sa contamination par les terres rares. La dissolution de l'hydroxyde de thorium dans l'acide nitrique est une méthode efficace car la solution qui en résulte peut être purifiée par extraction avec des solvants organiques.
Le thorium métallique (pur) est séparé de l'oxyde, du chlorure et du fluorure anhydres par réaction avec le calcium sous atmosphère inerte :
Le thorium est parfois extrait par électrolyse d'un fluorure dans un mélange de chlorure de sodium NaCl et de chlorure de potassium KCl de 700 à 800 °C dans un creuset en graphite. Le thorium hautement purifié peut être extrait à partir de son iodure par procédé Van-Arkel-de-Boer.
Extraction de l'uranium[modifier | modifier le code]
L'uranium est extrait de ses minerais de plusieurs façons. Une méthode consiste à brûler le minerai puis à le faire réagir avec de l'acide nitrique pour faire passer l'uranium en solution. En traitant cette solution avec une solution de phosphate de tributyle (CH3CH2CH2CH2O)3PO dans le kérosène conduit à la formation d'un composé organométallique de formule UO2(NO3)2((CH3CH2CH2CH2O)3PO)2. Les impuretés insolubles sont filtrées et l'uranium est extrait par réaction avec des hydroxydes sous forme de (NH4)2U2O7 ou avec du peroxyde d'hydrogène H2O2 sous forme de UO4·2H2O.
Lorsque le minerai d'uranium est riche en minéraux tels que la dolomite CaMg(CO3)2 ou la magnésite MgCO3, cette méthode consomme beaucoup d'acide. On préfère dans ce cas utiliser la méthode au carbonate pour en extraire l'uranium. Son ingrédient principal est une solution aqueuse de carbonate de sodium Na2CO3, qui convertit l'uranium en un complexe [UO2(CO3)3]4−, lequel est stable en solution aqueuse à faible concentration d'ions hydroxyde. L'avantage de cette méthode est que ses réactifs sont moins corrosifs que les nitrates et qu'elle fait précipiter la plupart des métaux autres que l'uranium. Son inconvénient est que les composés d'uranium tétravalent précipitent également. Pour cette raison, le minerai d'uranium est traité au carbonate de sodium à haute température et sous pression d'oxygène :
Cette équation suggère que le meilleur solvant pour le traitement du carbonate d'uranium est un mélange de carbonate CO32− et de bicarbonate HCO3−. À pH élevé, cela conduit à la précipitation de diuranate, qui est ensuite traité à l'hydrogène en présence de nickel pour donner un tétracarbonate d'uranium insoluble.
Un autre mode de séparation fait appel à des résines polymériques utilisées comme polyélectrolyte. Dans de tels dispositifs, l'uranium est séparé à l'aide de processus d'échange d'ions dans ces résines, puis est extrait de ces résines à l'aide de nitrate d'ammonium NH4NO3 ou d'acide nitrique HNO3 pour donner du nitrate d'uranyle UO2(NO3)2·6H2O. Ce dernier est alors chauffé pour donner du trioxyde d'uranium UO3, qui est réduit en dioxyde d'uranium UO2 avec l'hydrogène :
La réaction du dioxyde d'uranium avec l'acide fluorhydrique HF donne du tétrafluorure d'uranium UF4 :
Le tétrafluorure d'uranium donne de l'uranium métallique par réaction avec le magnésium.
Extraction du plutonium[modifier | modifier le code]
Afin d'extraire le plutonium du combustible nucléaire irradié, on commence par traiter l'uranium irradié aux neutrons avec de l'acide nitrique HNO3, puis un réactif comme le sulfate de fer(II) FeSO4 ou le peroxyde d'hydrogène H2O2 est ajouté à la solution pour agir comme réducteur en faisant ramener l'état d'oxydation du plutonium de +6 à +4 tandis que l'uranium demeure sous la forme de nitrate d'uranyle UO2(NO3)2. Les composés du plutonium(IV) sont enfin précipités sous l'action du carbonate d'ammonium (NH4)2CO3, qui fait remonter le pH jusqu'à 8.
Dans une autre méthode, le plutonium(IV) et l'uranyle UO22+ sont tout d'abord extraits par le phosphate de tributyle (CH3CH2CH2CH2O)3PO, puis sont mis à réagir avec l'hydrazine N2H4 pour récupérer le plutonium.
Applications[modifier | modifier le code]


.jpg)
Hormis l'uranium et le plutonium, utilisés par l'industrie nucléaire et pour la conception d'armes nucléaires, l'américium — notamment l'américium 241 — est utilisé dans les chambres d'ionisation des détecteurs de fumée, et le thorium a été utilisé dans les manchons à incandescence. L'utilisation comme combustible nucléaire tire parti de la très grande quantité d'énergie libérée lors de la fission de ces atomes, et de leur propriété de pouvoir entretenir une réaction en chaîne.
- Actinium — L'actinium 227 est employé comme source de neutrons. Son énergie spécifique élevée de 14,5 W g−1 et la possibilité d'en obtenir de grandes quantités en ont fait un isotope intéressant pour réaliser des générateurs thermoélectriques à radioisotope. L'actinium 228 est utilisé comme indicateur de radioactivité dans la mesure où il émet des électrons énergétiques de 2,28 MeV faciles à détecter. On utilise souvent des sources 228Ac-228Ra pour émettre des rayons γ à usage industriel ou médical.
- Thorium — Il est essentiellement utilisé dans la production de manchons à incandescence sous forme de dioxyde de thorium ThO2. Il trouve également un usage comme adjuvant dans certains alliages de magnésium et de zinc. Les alliages magnésium-thorium sont durs et légers avec un point de fusion et une ductilité élevés, ce qui en fait des matériaux utiles dans l'aéronautique et la production de missiles. Le thorium a également des propriétés intéressantes d'émission électronique, avec une grande durée de vie et une faible barrière de potentiel à l'émission. La teneur relative en isotopes de thorium et d'uranium est couramment utilisée pour estimer l'âge de divers matériaux dans le cadre des méthodes de datation par l'uranium-thorium, qui sont un cas particulier de datation radiométrique.
- Uranium — L'isotope le plus important pour la production d'énergie nucléaire est l'uranium 235. La fission d'un gramme d'uranium 235 libère environ 1 MW·jour. Cet isotope est présent dans le minerai d'uranium à hauteur de 0,72 % et est employé dans les réacteurs à neutrons thermiques. Cet isotope absorbe en effet les neutrons thermiques, déclenchant une réaction de fission au cours de laquelle d'autres neutrons sont émis, ce qui permet, lorsque la masse critique est atteinte, d'entretenir une réaction en chaîne. La fission d'un atome d'uranium consiste à faire éclater son noyau en deux fragments, avec libération de neutrons ; les fragments sont de nature variable, et le nombre de neutrons libérés également, par exemple :
- Le thorium 232 et l'uranium 233 sont également des isotopes potentiellement intéressants du point de vue de la production d'énergie nucléaire. L'émission de neutrons lors de la réaction de fission est importante non seulement pour entretenir la réaction en chaîne, mais également pour produire des nucléides plus lourds par capture neutronique suivie de désintégrations β−. L'uranium 239 se transmute ainsi en plutonium 239 par désintégrations β−, isotope également capable de fission spontanée. Le premier réacteur nucléaire a ainsi été construit non pas pour produire de l'énergie nucléaire mais pour produire du plutonium 239 à des fins militaires.
- Plutonium — L'utilisation de cet élément est avant tout militaire. Le plutonium 239 en est un composant essentiel dans la mesure où il est à la fois fissile et relativement facile à produire. Son utilisation permet de réduire la masse critique à environ le tiers de celle nécessaire avec l'uranium 235. Le plutonium 238 est potentiellement un isotope plus efficace pour les réacteurs nucléaires en raison de sa masse critique plus faible que celle de l'uranium 235, mais il continue de d'émettre de l'énergie thermique par désintégration même lorsque la réaction en chaîne est arrêtée par des barres de contrôle, et son coût de revient est relativement élevé. C'est la raison pour laquelle il est utilisé pour la réalisation de thermopiles pour l'astronautique et de générateurs thermoélectriques à radioisotopes pour l'exploration spatiale. Dans la mesure où sa désintégration produit des particules α relativement inoffensives et ne produit pas de rayons γ, cet isotope est utilisé comme source d'énergie pour pacemakers, où 160 mg assurent une durée d'alimentation cinq fois plus élevée que les piles conventionnelles.
Découverte et synthèse des actinides[modifier | modifier le code]
Voies de production[modifier | modifier le code]

Contrairement aux lanthanides, qui se rencontrent dans la nature en quantités appréciables (à l'exception du prométhium), la plupart des actinides sont des éléments très rares. Les éléments naturels les plus abondants sont le thorium et l'uranium ; et le plus facile à synthétiser est le plutonium ; les autres ne se rencontrent guère qu'à l'état de traces.
La possibilité d'éléments transuraniens a été suggérée par Enrico Fermi sur la base de ses expériences de 1934[11],[12]
Les transuraniens ne se trouvent pas en quantité significative dans la nature, et sont produits par réaction nucléaire. Il y a de nos jours deux grandes voies pour produire les isotopes au-delà du plutonium : l'irradiation par des flux de neutrons conduisant à une capture neutronique, ou l'irradiation par des faisceaux de particules, dans un accélérateur de particules.
La première voie est la principale pour les applications pratiques, la production d'actinides en quantité pondérale n'étant possible que par irradiation en réacteur nucléaire ; elle est toutefois limitée aux premiers éléments de la famille. Par exemple, dans des conditions d'irradiation neutronique d'un réacteur nucléaire, l'uranium 238 se transforme en partie en plutonium 239 : Les actinides de masses atomiques plus élevées sont synthétisés grâce à un accélérateur de particules en bombardant de l'uranium, du plutonium, du curium ou du californium avec des ions d'azote, d'oxygène, de carbone, de néon ou de bore. L'avantage de la seconde méthode est qu'elle permet de produire des éléments nettement plus lourds que le plutonium, ainsi que des isotopes présentant un déficit de neutrons. Ainsi, le nobélium a été produit en bombardant de l'uranium 238 avec du néon 22, suivant la réaction nucléaire : .
![{\displaystyle \mathrm {{}_{92}^{238}U+{}_{0}^{1}n\ {\xrightarrow {\ }}\ {}_{92}^{239}U\ {\xrightarrow[{23.5\ min}]{\beta ^{-}}}\ {}_{93}^{239}Np\ {\xrightarrow[{2.3\ jours}]{\beta ^{-}}}\ {}_{94}^{239}Pu\ {\xrightarrow[{2.4\cdot 10^{4}\ ans}]{\alpha }}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/bb194337a1b8571d962f0526fc5489d417049a3e)

Entre 1962 et 1966, les États-Unis conduisirent une série de six essais nucléaires souterrains pour essayer d'analyser la production des isotopes lourds dans un contexte de haut flux neutronique. De petits échantillons de roche furent prélevés immédiatement après l'explosion pour analyser les produits de l'explosion, mais aucun isotope de masse atomique supérieure à 257 ne fut jamais identifié, bien que la théorie prévoyait à l'époque, dans cette région, un îlot de stabilité d'isotopes ayant une période radioactive relativement longue en radioactivité alpha.
Découverte des actinides naturels[modifier | modifier le code]
| N° | Nom | UICPA | Isotope découvert |
Année de découverte |
Méthode de découverte |
|---|---|---|---|---|---|
| 89 | Actinium | Ac | naturel | 1899 | Séparation chimique |
| 90 | Thorium | Th | naturel | 1829 | Séparation chimique |
| 91 | Protactinium | Pa | 234 mPa | 1913 | Séparation chimique |
| 92 | Uranium | U | naturel | 1789 | Séparation chimique |
| 93 | Neptunium | Np | 239neptunium | 1940 | Bombardement de 238U par des neutrons lents |
| 94 | Plutonium | Pu | 238plutonium | 1941 | Bombardement de 238U par des deutons |
| 95 | Américium | Am | 241américium | 1944 | Bombardement de 239Pu par des neutrons |
| 96 | Curium | Cm | 242curium | 1944 | Bombardement de 239Pu par des particules α |
| 97 | Berkélium | Bk | 243berkélium | 1949 | Bombardement de 241Am par des particules α |
| 98 | Californium | Cf | 245californium | 1950 | Bombardement de 242Cm par des particules α |
| 99 | Einsteinium | Es | einsteinium | 1952 | En tant que produit de l'explosion nucléaire Ivy Mike |
| 100 | Fermium | Fm | 255fermium | 1952 | En tant que produit de l'explosion nucléaire Ivy Mike |
| 101 | Mendélévium | Md | 256mendélévium | 1955 | Bombardement de 253Es par des particules α |
| 102 | Nobélium | No | 256nobélium | 1965 | Bombardement de 243Am par de l'15N ou bombardement de 238U par des particules α ou bombardement de 238U par du 22Ne |
| 103 | Lawrencium | Lr | 258lawrencium | 1961–1971 | Bombardement de 252Cf par du 10B ou du 11B et de 243Am par de l' 18O |
- L'uranium a été isolé en 1789 par le chimiste allemand Martin Heinrich Klaproth à partir de minerai de pechblende. Il baptisa cette matière « uranium » en référence à la planète Uranus, qui avait été découverte huit années plus tôt. Klapoth obtint un précipité jaune (probablement du diuranate de sodium) par dissolution de la pechblende dans de l'acide nitrique, puis en neutralisant la solution obtenue par de l'hydroxyde de sodium. Il réduisit alors ce précipité jaune avec du charbon de bois, et obtint une substance noirâtre qu'il prit pour un métal[14]. Ce n'est que soixante ans plus tard que le chimiste français Eugène-Melchior Péligot analysa cette poudre comme de l'oxyde d'uranium. Il isola également le premier échantillon métallique d'uranium, en chauffant du tétrachlorure d'uranium en présence de potassium[15]. La masse atomique de l'uranium était alors estimée à 120, mais en 1872, Dmitri Mendeleïev révisa cette valeur à 240 en se fondant sur son tableau périodique des éléments. Par la suite, cette valeur fut confirmée expérimentalement en 1882 par K. Zimmerman[16],[17].
- L'oxyde de thorium a été découvert par Friedrich Wöhler dans un minerai de Norvège en 1827. Ce minerai fut analysé par Jöns Jacob Berzelius en 1828. Par réduction du tétrachlorure de thorium avec du potassium, il obtint un métal qu'il appela thorium, en référence à Thor, dieu du tonnerre et des éclairs de la mythologie nordique[18],[19]. C'est cette même méthode qui fut par la suite utilisée par Pélogot pour isoler l'uranium.
- L'actinium a été découvert en 1899 par André-Louis Debierne, un assistant de Marie Curie, dans les restes de pechblende qui avaient été traités pour en extraire le radium et le polonium. Il décrivit initialement cette substance comme similaire au titane[20] puis (en 1900) comme similaire au thorium[21]. Cette découverte de l'actinium par Devierne a cependant été remise en cause en 1971[22] et en 2000[23], sur la base de ce que les travaux publiés par Devbierne en 1904 contredisaient ses publications précédentes de 1899 et 1900. Le terme «actinium» vient du grec aktis, aktinos (ακτίς, ακτίνος) qui signifie un rayon lumineux. Du fait de sa similarité avec le lanthane et de sa très faible abondance, l'actinium ne fut isolé à l'état pur qu'en 1950.
- Le protactinium fut peut-être isolé en 1900 par William Crookes[24]. Il fut identifié en 1913, quand Kazimierz Fajans et Oswald Helmuth Göhring (es) identifièrent son isotope 234 mPa (d'une demi-vie de 1,17 minute) dans leur étude de la chaîne de désintégration de 238U. Ils appelèrent initialement ce nouvel élément « brévium » (du latin brevis, d'une courte durée). En 1918 deux groupes de scientifiques conduits par l'Autrichienne Lise Meitner, l'Allemand Otto Hahn et l'Anglais John Cranston (en) firent indépendamment la découverte de l'isotope 231Pa, et lui donnèrent le nom de protoactinium (du grec πρῶτος / prỗtos, « premier » et ἀκτίς / ἀκτίς, « rayon » : le premier des rayonnants), terme abrégé en protactinium en 1949. Cet élément ne fut guère caractérisé avant 1960, date à laquelle A. G. Maddock et ses collaborateurs parvinrent à produire 130 grammes de protactinium à partir de 60 tonnes de déchets de minerai d'uranium.
Synthèse des actinides artificiels[modifier | modifier le code]

La famille des actinides transuraniens a été découverte aux débuts de la physique nucléaire, entre les années 1940 et 1960.
- Le neptunium fut découvert par Edwin McMillan et Philip H. Abelson en 1940 à Berkeley. Ils produisirent l'isotope 239Np (d'une demi-vie de 2,4 jours) en bombardant de l'uranium avec des neutrons lents. Ils baptisèrent cette substance « neptunium » en référence à la planète Neptune, la planète suivant Uranus qui avait donné son nom à l'uranium. Ce fut le premier transuranien produit artificiellement.
- L'isotope 238Pu a été produit le et caractérisé en 1941 par l'équipe de Glenn Seaborg en bombardant une cible d'uranium par du deutérium au cyclotron de Berkeley[25].
- Le curium a été découvert en été 1944 par Glenn T. Seaborg et ses collègues Ralph A. James et Albert Ghiorso. Dans leur série d'expériences, ils ont utilisé le cyclotron de 60 pouces de l'université de Californie à Berkeley.
- L'américium 241 fut synthétisé pour la première fois par Glenn T. Seaborg, Leon Morgan (es), Ralph James, et Albert Ghiorso vers la fin de l’année 1944 au laboratoire métallurgique de l’université de Chicago (connu maintenant sous le nom d’Argonne National Laboratory), par l'irradiation de plutonium dans un réacteur nucléaire. L’américium a été nommé en référence au continent américain, par analogie avec l’europium, élément de la famille des lanthanides dont il est l’homologue chimique.
- Le berkélium a été synthétisé pour la première fois en décembre 1949 par Stanley G. Thompson (en), Glenn T. Seaborg, Kenneth Street, Jr. (en), et Albert Ghiorso à l'université de Californie à Berkeley. Le premier isotope produit a un nombre de masse de 243 et se dégrade avec une demi-vie de 4,5 heures.
- Le californium 245 a été produit pour la première fois en 1950 par Stanley G. Thompson (en), Glenn T. Seaborg, Kenneth Street, Jr. (en), et Albert Ghiorso à Berkeley, en Californie, d'où son nom : il avait alors été obtenu en bombardant une cible de curium 242 avec un faisceau de particules α pour produire du 245Cf par une réaction (α, n).
- L'einsteinium, nommé d'après Albert Einstein, et le fermium, nommé d'après Enrico Fermi, ont été découverts en 1952 par Albert Ghiorso dans les débris résultant de Ivy Mike, la première explosion thermonucléaire. Le 255Fm avait été créé par combinaison de l'uranium 238 et de 17 neutrons sous l'effet du flux neutronique intense. Ces résultats avaient initialement été classifiés, mais l'équipe de Berkeley parvint à synthétiser ces deux éléments par des moyens civils en soumettant du plutonium 239 à un bombardement neutronique, et publièrent ce résultat en 1954, signalant que ce n'était pas la première découverte de ces éléments. Les études des retombées de Ivy Mike furent finalement déclassifiées en 1955.
- Le mendélévium fut obtenu pour la première fois sous forme de 256Md](d'une demi-vie de 87 minutes) par Albert Ghiorso, Glenn T. Seaborg, Gregory R. Choppin, Bernard G. Harvey et Stanley G. Thompson, en bombardant une cible de 253Es par un faisceau de particules alpha. Ce fut le premier élément à être synthétisé un atome à la fois.
- Le nobélium fut annoncé en 1957 par l'Institut Nobel de physique à Stockholm, et plus tard par Albert Ghiorso, Torbjorn Sikkeland, J.R.Walton et Glenn Seaborg (États-Unis) en 1958. Il fut nommé nobélium en l'honneur d'Alfred Nobel. Il a été identifié pour la première fois avec certitude en 1965 sous forme de 256No par le Russe Gueorgui Fliorov et son équipe, en bombardant de l'uranium 238 avec du néon 22.
- Le lawrencium, dernier des actinides, a été observé pour la première fois en par Albert Ghiorso, Torbjørn Sikkeland, Almon E. Larsh et Robert M. Latimer (États-Unis), au Laboratoire national Lawrence-Berkeley de l'université de Californie à Berkeley, en bombardant une cible de trois isotopes de californium par des ions de bore 10 et de bore 11 pour produire du 258Lr. Il porte le nom d'Ernest Orlando Lawrence, qui découvrit le principe du cyclotron en 1929.
Identification comme famille d'éléments[modifier | modifier le code]
De même que les lanthanides, les actinides forment une famille d'éléments aux propriétés chimiques semblables.
Toutefois, bien que quatre actinides étaient déjà connus dans les années 1930, le fait qu'ils puissent former une famille comparable aux lanthanides n'avait pas encore été compris. Le point de vue prédominant à l'époque était qu'ils formaient une suite régulière d'éléments de la septième période, dans laquelle thorium, protactinium et uranium avaient sur la sixième période le hafnium, le tantale et le tungstène comme analogues respectifs. C'est probablement Victor Goldschmidt qui introduisit le terme « actinide » en 1937[26],[27].
Mais la synthèse des transuraniens renversa progressivement cette vision des choses. En 1944, le constat que le curium ne présente pas de degré d'oxydation au-dessus de 4 (alors que son analogue supposé, le platine, peut atteindre un degré d'oxydation de 7) conduisit Glenn Seaborg à formuler l'hypothèse d'une famille d'actinides. L'étude des actinides déjà isolés et la découverte d'autres éléments transuraniens confirma l'existence de cette famille de transition.
Production d'actinides dans les réacteurs nucléaires[modifier | modifier le code]
Captures neutroniques[modifier | modifier le code]

Les actinides artificiels qui présentent un intérêt pratique sont les noyaux lourds (isotopes) formés dans les réacteurs par capture successive de neutrons par les noyaux du combustible.
Lors de l'irradiation en réacteur, les atomes d'actinide présents dans le combustible peuvent capturer un neutron sans subir de fission. C'est le cas notamment de l'isotope 238 de l'uranium, qui n'est pas fissile en spectre thermique ; mais tous les actinides présents présentent une section efficace de capture neutronique. La vitesse de transmutation en réacteur dépend de la valeur de cette section efficace. Pour un flux neutronique en réacteur typique de l'ordre de 1 × 1014 n cm−2 s−1, une section efficace de l'ordre de σ=1 barn (soit 1 × 10−24 cm2) aura en un an (soit 3,156 × 107 s) une déplétion de : 1 × 1014 n cm−2 s−1 × 1 × 10−24 cm2 × 3,156 × 107 s = 0,316 %
Le barn est l'ordre de grandeur de la section efficace de capture de l'238U, soit 2,68 barns en neutrons thermique : au bout d'un an en réacteur, près de 1 % (2,68 x 0,316 = 0,846 %) de l'uranium aura été transformé en plutonium. Cependant, ce calcul qui assimile la « déplétion » à une probabilité de capture n'est correct qu'en première approximation, lorsque cette probabilité est faible ; la probabilité réelle d'avoir subi la réaction suit en réalité non pas une loi linéaire mais une loi exponentielle, saturant asymptotiquement à 100 %. Ainsi, une section efficace de 1000 barns conduira dans les mêmes conditions à calculer une « déplétion » mille fois plus importante, mais en termes de probabilité la part effectivement touchée n'est évidemment pas un absurde 315,6 %, mais : 1-exp(-3.156) = 95,74 % La demi-vie en réacteur d'un tel isotope est la durée pour laquelle cette « déplétion » vaut Log(2)=69,31 %. Cette demi-vie est donc inversement proportionnelle à la section efficace. Si dans l'exemple ci-dessus la « déplétion » est de 3.156 par an, la moitié de l'isotope sera consommé en Log(2)/3.156 ans = 0.22 ans = 80 jours.
Ces captures, le plus souvent suivies de décroissance radioactive bêta moins, conduisent à une augmentation du numéro atomique (du nombre de protons contenus dans le noyau). À partir de l'uranium initial il se forme alors des transuraniens : d'abord du plutonium, puis des actinides mineurs : principalement le neptunium (237) (produit d'une part par capture de l'uranium 236 formé à partir de l'uranium 235 –environ 20,3 % des fissions et 16,8 % des captures– et d'autre part par réaction (n,2n) sur l'uranium 238), l’américium (241, 243) et le curium (243, 244, 245).
Les isotopes des éléments transuraniens ont souvent une demi-vie très courte. Les actinides à demi-vie très courte présentent un excès de neutrons, qu'ils résolvent rapidement (avec des demi-vies de l'ordre de la journée) par radioactivité bêta moins, par transformation d'un neutron en un proton (qui augmente d'une unité le numéro atomique) et d'un électron expulsé du noyau.
Quelques isotopes de Np, Pu, Am et Cm sont relativement plus stables, et sont produits en quantités pondérables dans les réacteurs nucléaires. Les principaux sont le plutonium, le neptunium 237(représente à lui seul près de 50 % des actinides mineurs formés), les américium 241 et 243 et les curium 244 et 245 (les proportions typiques sont données plus loin). Ils ont généralement une radioactivité alpha, avec une demi-vie qui peut aller de quelques dizaines années pour les curiums 243 et 244 à 2,144 millions d'années pour le plus stable, le neptunium 237.
Ce sont ces actinides que l'on peut retrouver comme sous-produits d'un réacteur nucléaire. Même s'ils ne sont pas nécessairement fissiles en neutrons thermiques, ils sont tous fissibles avec une section efficace de 0,5 à 2 barn avec des neutrons d'énergie > 2 MeV. Ils peuvent donc être détruits en réacteur à neutrons rapides, ou peuvent être considérés comme déchets définitifs et stockés comme déchets nucléaires HAVL.
Équilibre neutronique des réacteurs[modifier | modifier le code]
Dans un réacteur nucléaire, une réaction nucléaire ne peut fonctionner de manière auto-entretenue que si les neutrons produits par la fission d'un atome (généralement de deux et demi à trois en moyenne) ne subissent pas trop de pertes avant de contribuer à une nouvelle fission. Outre les pertes par diffusion et par activation des constituants du réacteur, les neutrons sont également consommés quand il y a une capture neutronique par un noyau d'actinide. De ce fait, la capacité des actinides à absorber des neutrons en fait en première approche un poison neutronique : plus il y en a dans le cœur du réacteur, et plus la réactivité du cœur sera compromise. Si on laisse trop d'actinides dans le cœur d'un réacteur nucléaire, il peut à terme ne plus fonctionner.
En deuxième approche, l'existence d'actinides fissiles viennent nuancer ce bilan, en ce qui concerne les radionucléides fertiles. Si par exemple la capture neutronique faite par un atome de 238U fait perdre un neutron dans l'équilibre neutronique du cœur sans bénéfice immédiat, elle transforme aussi à terme cet atome en un atome de 239Pu fissile. À plus longue échéance, un second neutron pourra faire fissionner ce dernier et produire les « deux et demi à trois en moyenne » neutrons associés à cette dernière fission. La capture d'un neutron, dans ce cas, conduit donc à un déficit immédiat sur la réactivité, mais le bilan neutronique global reste en moyenne légèrement positif : globalement, deux neutrons ajoutés à l'atome de 238U auront produit « deux et demi à trois en moyenne » neutrons nouveaux, ce qui ne compromet pas fondamentalement la possibilité d'une réaction en chaîne.
En revanche, si l'isotope produit n'est pas un isotope fertile, le bilan neutronique est nécessairement négatif : il faudra au moins une capture neutronique supplémentaire pour conduire à une fission ; et le bilan global sera au mieux de trois neutrons pour une fission, qui ne produit que « deux et demi à trois en moyenne » neutrons nouveaux : la présence de cet isotope aura été une charge pour l'équilibre neutronique du cœur.
La charge des actinides sur le bilan neutronique est d'autant pire que le nombre de neutrons à absorber avant d'atteindre un isotope fissile sera important.
Pour les actinides supérieurs (berkélium et curium), les captures neutroniques successives conduisent à des radioisotopes fortement radioactifs en radioactivité alpha, qui émettent un noyau d'hélium, éventuellement avant d'avoir eu le temps de fissionner. Dans ce cas, le bilan neutronique est encore plus noir : l'émission d'une particule alpha signifie que globalement quatre neutrons ont été absorbés (dont deux transformés en protons) sans produire de fission, et le noyau est retourné au stade où il était quatre captures neutroniques en amont : si un noyau suit un tel cycle, quatre neutrons auront été consommés en perte sèche pour ce qui est de l'entretien de la réaction nucléaire.
Cet impact des actinides sur le bilan neutronique est surtout important pour les réacteurs modérés. Dans le cas des réacteurs à neutrons rapides, les actinides formés sont tous plus ou moins fissibles ; ils sont donc plus rapidement consommés par le flux neutronique, et une capture neutronique éventuelle conduit directement à un autre atome fissible, comme c'est le cas pour les noyaux fertiles.
Actinides du cycle du thorium[modifier | modifier le code]
| 230Th 4 | → | 231Th | ← | 232Th 9 | → | 233Th | (En blanc : t½<27 j) | |||||||
| ↓ | ↓ | |||||||||||||
| 231Pa 4 | → | 232Pa | ← | 233Pa | → | 234Pa | (Coloré : t½>68 a) | |||||||
| ↑ | ↓ | ↓ | ↓ | |||||||||||
| 231U | ← | 232U 1 | ↔ | 233U 5 | ↔ | 234U 5 | ↔ | 235U 9 | ↔ | 236U 7 | → | 237U | ||
| ↓ | ↓ | ↓ | ↓ | |||||||||||
| (Produits de fissions à t½<90 a ou t½>200 ka) | 237Np 6 | |||||||||||||
Dans le cas d'un réacteur fonctionnant sur le cycle du thorium, l'actinide initial est formé par le thorium 232, qui est un isotope fertile.
- 232Th capte un neutron (σ=7.3b) pour devenir 233Th. Celui-ci émet rapidement (T/2=22,3 min) un électron (et un neutrino) par l'émission bêta, pour se muer en protactinium 233, un premier actinide mineur ; et dans une seconde émission bêta (T/2=27 j), 233Pa devient de l'uranium 233, le combustible.
- 233U est la matière fissile de ce cycle (σ=530b). Cependant, dans un peu moins de 10 % des cas[28] il peut absorber à son tour un neutron (σ=47b), pour former du 234U fertile.
- 234U est relativement stable (T/2=245 500 ans) et est un isotope fertile. Par une capture supplémentaire (σ=98b), il finit généralement par former du 235U, qui est la matière fissile du cycle de l'uranium.
Le cycle du thorium ne peut être envisagé que dans le cadre d'un cycle surgénérateur, où le bilan neutronique permet de créer la matière fissile qui alimentera le cycle. Dans cette production de la matière fissile, une petite partie (10 % des cas) est perdue pour ce cycle, mais se retrouvera dans le cycle de l'uranium : la perte d'un neutron ne compte donc ici que pour 10 %, le devenir du radionucléide étant discuté au cycle suivant.
Outre les captures neutroniques, dans le cas du thorium les réactions (n,2n) sont importantes par leurs conséquences. De telles réactions sont l'inverse d'une capture : le neutron incident quand il est suffisamment énergétique fait en quelque sorte un « carreau » (comme à la pétanque) et déloge du noyau un neutron supplémentaire, faisant baisser d'une unité son poids atomique.
Ce phénomène peut intervenir dans deux points du cycle du thorium, suivant que l'expulsion intervient avant ou après une première capture :
- initialement, un atome de 232Th peut par réaction (n,2n) sur des neutrons de plus de 6,4 MeV se transformer en 231Th, qui se transforme rapidement en 231Pa. Dans un deuxième temps, celui-ci pourra absorber un neutron supplémentaire, formant du 232Pa qui se transforme rapidement en 232U ;
- alternativement, après la première capture neutronique par le thorium, le 233Pa du cycle principal peut par réaction (n,2n) sur des neutrons de plus de 6,6 MeV former du 232Pa, qui finit de même en 232U.
Cet uranium 232, faiblement fissile et fertile (σ~74b), atteint assez rapidement son équilibre séculaire et accompagne donc à l'état de trace l'uranium 233 normalement formé par le cycle.
Ce marquage isotopique de l'uranium 233 est important, parce que la chaîne de désintégration de 232U comprend un émetteur gamma très énergétique, donc très pénétrant. D'autre part, ses descendants sont tous de demi-vie très brève, l'équilibre séculaire avec ces émetteurs gamma est donc très rapidement atteint. Enfin, la demi-vie T/2= 68,9 ans de 232U le rend à la fois fortement radioactif, et très persistant à l'échelle de temps historique (sa radioactivité n'aura diminué d'un facteur mille qu'au bout de sept siècles).
Ce rayonnement impose des protections radiologiques importantes dans toutes les opérations concernant l'uranium produit par ce cycle, même quand il a été isolé des produits de fission et des autres actinides, ce qui rend ces opérations techniquement plus complexes et économiquement plus couteuses. Cet inconvénient est au contraire un avantage en ce qui concerne la lutte contre la prolifération, parce que le rayonnement gamma produit par cet uranium est très facile à détecter, ce qui rend impossible toute dissimulation de cette matière à des contrôles officiels.
Actinides du cycle de l'uranium[modifier | modifier le code]
Le cycle de l'uranium se fonde sur la fission de l'uranium 235.
- 235U est une matière fissile, mais dans environ un cas sur six, capte un neutron sans fissionner pour former 236U, quasi stable (T½ = 23,42 Ma).
- 236U laissé en irradiation finit par former 237U, qui se transforme rapidement (T½ = 6,75 j) en neptunium 237, quasi stable (T½ = 2,144 Ma).
- 237Np capture un neutron (σ = 176 barns) pour former 238Np, lequel se transforme rapidement (T½ = 2,1 j) en plutonium 238, fortement radioactif (T½ = 87,75 ans).
- 238Pu est faiblement fissile, et si l'on poursuit l'irradiation, se transformera dans plus de 90 % des cas en plutonium 239, où il rejoint le cycle du plutonium.
Pour être complet sur le cycle de l'uranium, il convient de noter que l'uranium 238, base du cycle du plutonium, peut également perdre un neutron par une réaction (n,2n). Il se transforme alors en 237U puis en 237Np.
Le neptunium est l'actinide important de ce cycle. 237Np peut être séparé chimiquement des combustibles irradiés, puis remis en irradiation dans des cibles d'irradiation pour produire du plutonium 238, dont il est à nouveau séparé chimiquement. Ce 238Pu peut ainsi être obtenu sans être mélangé à 239Pu issu de l'irradiation de 238U présent dans le combustible initial. Il sert principalement pour la fabrication de générateur thermoélectrique à radioisotope.
Actinides du cycle du plutonium[modifier | modifier le code]

Dans le cas d'un réacteur fonctionnant sur le cycle du plutonium, l'actinide initial est formé par l'uranium 238, qui est un isotope fertile.
- 238U capte parfois un neutron pour former du 239U, lequel se transforme rapidement (T½ = 24 min) en neptunium 239, puis (T½ = 2,3 jours) en plutonium 239, la principale matière fissile de ce cycle. C'est essentiellement l'intégrale de résonance qui contribue à cette absorption (σ = 275,6 b à 300 K), la capture de neutrons thermiques étant comparativement négligeable (σ = 2,69 b)[29]. L'intégrale de résonance augmentant avec la température, ce phénomène introduit une rétroaction négative stabilisant le fonctionnement des réacteurs nucléaires.
- 239Pu est une matière assez stable (T½ = 24 110 ans) et fissile (σ = 790 b), mais une fois sur trois (σ = 271 b)[30] il capture le neutron incident pour former du 240Pu.
- 240Pu, stable en réacteur (T½ = 6 563 ans), est fertile (σ = 297,2 b)[31] : sous irradiation il peut capturer un neutron pour former du 241Pu fissile.
- 241Pu est une matière fortement radioactive (T½ = 14,35 ans) et fissile (σ = 1,060 kb)[32]. Sous irradiation, il peut néanmoins capturer un neutron une fois sur quatre (σ = 373,7 b)[32] et se transformer en 242Pu.
- 242Pu est stable (T½ = 373 000 ans). En capturant parfois un neutron (σ = 20,09 b)[33] il se transforme en 243Pu, très instable, qui se transforme rapidement (T½ = 5 h) en americium 243.
Le plutonium formé par ce cycle, qui peut être extrait chimiquement, est un mélange isotopique contenant au départ surtout du 239Pu, et d'autant plus d'isotopes plus lourds (240, 241 et 242) que l'irradiation aura été prolongée longtemps.
La principale caractéristique du plutonium, par contraste avec l'uranium naturel, est d'être naturellement enrichi en isotopes fissiles : la mise en œuvre d'une « simple » séparation chimique est suffisante pour obtenir de la matière fissile, sans avoir besoin de recourir à une séparation isotopique. Cela en fait la principale matière première nécessaire pour alimenter un cycle surgénérateur, en concurrence avec l'uranium fortement enrichi, pour les projets de réacteurs de quatrième génération. C'est également parce que cette matière fissile est d'obtention comparativement plus facile que l'uranium hautement enrichi que la première explosion nucléaire a été réalisée avec du plutonium, et que la prolifération nucléaire passe le plus souvent par le détournement à des fins militaires du plutonium produit dans des réacteurs nucléaires réputés civils, mais hors contrôle de l'AIEA.
La série du plutonium formé en réacteur s'arrête en pratique au 242Pu du fait de la très forte instabilité du 243Pu, qui dans le flux neutronique relativement limité des réacteurs, se désintègre (T½ = 5 h) statistiquement bien avant d'avoir pu capturer un neutron supplémentaire (T½ de l'ordre de quelques dizaines d'années en réacteur), ce qui aurait formé du 244Pu. Paradoxalement, donc, le plutonium 244, seul isotope suffisamment stable pour être présent à l'état de traces dans la nature, est pratiquement absent du plutonium formé en réacteur. Sa formation naturelle est due aux très hauts flux neutroniques rencontrés dans le processus r de l'explosion des supernovæ ; et on en retrouve également des traces dans les isotopes formés lors d'une explosion atomique.
En dehors des irradiations très brèves conçues pour produire essentiellement du 239Pu, et donc destinées à un usage militaire, le plutonium formé comprendra toujours une fraction significative de 241Pu. La formation de ce plutonium s'accompagne alors d'une faible production d'américium, qui le rend à terme très fortement irradiant à cause de l'isotope 241Am. En attendant suffisamment longtemps, la radioactivité bêta de 241Pu en transformera une fraction en américium 241 (T½ = 14,35 ans), première étape de sa chaîne de désintégration. Dans 85 % des cas, la désintégration α se produit par une émission d'une particule de 5,485 MeV vers un état excité du 237Np, qui libère ensuite un rayon gamma de 59,54 KeV pour revenir à son fondamental. Le spectre énergétique de la désintégration de l'américium 241 étant cependant complexe avec de nombreuses transitions différentes possibles, il génère au total plus de 200 raies d'émissions alpha, gamma et X[34].
- 241Am est très faiblement fissile (σ = 3,122 b)[35] ; en réacteur c'est surtout un poison neutronique qui tend à capturer un neutron (σ = 691,4 b)[35] pour former du 242Am à 90 % et son isomère 242mAm à 10 %[36]. Bien que formé majoritairement, 242Am est très fortement radioactif (T½ = 16 h) et forme rapidement du 242Cm (à 83 %) ou du 242Pu.
- 242mAm sous irradiation en réacteur est un isotope qui disparaît rapidement. Il est facilement fissile (σ = 7,028 kb)[37], mais une fois sur six ou sept, il capture un neutron supplémentaire pour former du 243Am (σ = 1,258 kb)[37].
- 243Am provient donc d'une capture neutronique effectuée soit par 242mAm, soit par 242Pu. C'est le plus stable des isotopes de l'américium, avec une demi-vie de 7 370 ans. C'est également un poison neutronique mineur, capturant un neutron pour former du 244Am (σ = 80,83 b)[38], lequel se désintègre très rapidement (T½ = 10,1 h) pour former du 244Cm.
L'américium que l'on peut séparer par voie chimique a donc une composition isotopique potentiellement très variable. Le point d'entrée dans la production de l'américium est donc le 241Pu, qui résulte d'une irradiation prolongée en réacteur. À partir de cet isotope, le temps produira de l'241Am à partir du moment où l'âge du 241Pu formé est une fraction significative de sa demi-vie (T½ = 14,35 ans) ; une irradiation prolongée de 241Pu produira du 242Pu puis du 243Am ; et une irradiation prolongée de 241Pu âgé produira une fraction significative de 242mAm.
S'agissant du plutonium, la production continue d'241Am complique fortement son utilisation par les mesures de radioprotection qu'il impose, d'autant plus nécessaire que le plutonium sera plus âgé : sa radioactivité croît fortement avec le temps, jusqu'à atteindre son équilibre séculaire en une cinquantaine d'années. Il est possible d'éliminer l'américium par voie chimique, le rendant temporairement peu irradiant, et ce plutonium « frais » peut alors être employé avec des contraintes de radioprotection beaucoup plus faible. Mais seule la fraction de 241Pu déjà transformée en 241Am peut être ainsi éliminée, le reliquat continuant à produire du 241Am en permanence. Cet état faiblement irradiant ne dure donc pas, tant que la fraction de 241Pu reste significative dans le mélange. Avec une demi-vie de 14,35 ans, il faut donc attendre quelques siècles pour que la séparation de l'américium permette d'obtenir du plutonium à la fois faiblement irradiant, et le restant à long terme.
Actinides mineurs ultérieurs[modifier | modifier le code]
L'entrée dans la série du curium peut se faire par deux points :
- l'américium 241 peut capturer un neutron pour se transformer en 242Am, puis rapidement (T/2=16h) en curium 242, ou en 242Pu du cycle précédent ;
- l'américium 243 peut capturer un neutron pour se transformer en 244Am, puis rapidement (T/2=10h) en curium 244.

Une fois atteint le curium, les captures neutroniques successives vont augmenter la masse du noyau depuis 242Cm jusqu'à 249Cm.
- Dans cette série, les premiers éléments (242 à 244) sont fortement radioactifs, et retransforment les noyaux en plutonium par une radioactivité alpha.
- Dans cette série, les éléments 243Cm, 245Cm et 246Cm sont fissiles.
À partir de 245Cm, les demi-vies sont supérieures au millier d'années, et la voie d'évolution principale sous irradiation sera soit la fission, soit l'accumulation de neutrons jusqu'au 248Cm.
- 248Cm peut capturer un neutron et se transformer en 249Cm, qui se transforme rapidement en berkélium 249.
- 249Bk peut se désintégrer en 249Cf, ou capturer un autre neutron pour se transformer en 250Bk qui se désintègre rapidement en 250Cf.
Les radionucléides de californium sont assez fortement radioactifs. Ils peuvent soit accumuler encore des neutrons, passant de 249Cf à 252Cf, soit subir une désintégration alpha qui les fera retomber sur la série du curium.
L'accumulation des neutrons plafonne en pratique sur le californium 253, très instable, qui subit rapidement une désintégration alpha suivie d'une bêta, fait retomber l'isotope sur le même cycle : 253Cf ⇒ 249Cm ⇒ 249Bk. Le cycle peut alors repartir pour quatre absorption neutronique supplémentaires produisant à chaque fois un noyau d'hélium.
- Problématique de l'élimination
Des études et expérimentions ont été conduites pour évaluer les possibilités de transmutation en réacteur de ces éléments, d'une manière qui privilégie la fission sur la capture neutronique. Si la capture neutronique est trop importante, on tombe en effet sur les cycles supérieurs décrits ci-dessus.
Le bilan neutronique étant toujours une question importante, la bonne façon d'éliminer les actinides mineurs est donc de les fissionner le plus rapidement possible, et pour ce faire, d'utiliser un réacteurs à neutrons rapides voire un réacteur nucléaire piloté par accélérateur.
Actinides mineurs et déchets nucléaires[modifier | modifier le code]
Généralités[modifier | modifier le code]

La masse totale d'actinides mineurs formés dans le combustible retraité issu des réacteurs à eau pressurisée (taux de combustion moyen de 33 000 à 45 000 MWj/tMLi[39]) varie en fonction du taux de combustion et du type de combustible utilisé (uranium naturel enrichi ou MOX ou URE) entre 2,7 et 3,2 % de la masse de produits de fission formés. On peut donc voir que la « perte » en atomes lourds occasionnée par la mise au déchet des actinides mineurs ne dépasse pas 3,5 % de la ressource totale en uranium.
La composition massique typique des actinides mineurs dans le combustible retraité (33 000 à 45 000 MWj/tMLi[39]) cinq ans après déchargement du réacteur est donnée ci-après. Pour les corps à période inférieure à mille ans, le tableau donne le premier isotope à vie très longue (supérieure à mille ans) rencontré dans la décroissance vers la situation stable (le plomb dans la majorité des cas).
| Corps | Période | % mini | % maxi | 1er descendant à vie très longue |
Période du descendant | Observation |
|---|---|---|---|---|---|---|
| Cm 242 | 162,19 j | 0,01 | 0,03 | U 234 | 245,5 ka | en passant par 238Pu |
| Total vie très courte | < 1 a | 0,01 | 0,03 | |||
| Cm 244 | 18,1 a | 2,50 | 4,00 | Pu 240 | 6,56 ka | |
| Cm 243 | 29,1 a | 0,03 | 0,05 | Pu 239 | 24,1 ka | |
| Total vie moyenne | 1 a < < 31 a | 2,53 | 4,05 | |||
| Np 237 | 2,144 Ma | 45,00 | 55,00 | |||
| Am 241 | 432,2 a | 30,00 | 33,00 | Np 237 | 2,144 Ma | |
| Am 243 | 7,37 ka | 12,00 | 14,00 | |||
| Cm 245 | 8,5 ka | 0,15 | 0,20 | |||
| Am 242m | 141 a | 0,08 | 0,12 | U 234 | 245,5 ka | |
| Cm 246 | 4,73 ka | 0,02 | 0,04 | |||
| Total vie longue | > 31 a | 87,25 | 100,0 | |||
| Total général | sans objet | 100,0 | 100,0 | 2,7 à 3,2 % des PF |
Tous ces éléments, notamment ceux à vie courte et moyenne, contribuent fortement au dégagement thermique du combustible irradié et des déchets. Ils sont tous émetteur alpha ou ont des descendants qui le sont[40] et produisent donc de l'hélium.
Risques et dangers liés aux actinides mineurs (AMin)[modifier | modifier le code]
À l'usine de retraitement les AMin, à l'état chimique d'oxydes, se retrouvent mélangés aux produits de fission (PF). Incorporés dans un verre, ils constituent une partie des déchets de type C (HAVL)[41] De façon générale, ils représentent les déchets radioactifs qui posent les principaux problèmes, notamment au niveau du stockage des déchets radioactifs en couche géologique profonde pour les raisons suivantes :
- en tant qu'émetteurs alpha, ils présentent une forte toxicité chimique et radiologique si l'élément entre dans la chaîne alimentaire (inhalé, injecté ou ingéré) ;
- ils dégagent une forte chaleur ; les émissions alpha des actinides sont toutes à une énergie élevée de l'ordre de 4,5 à 5,5 MeV ;
- ils dégagent de l'hélium, qui est suspecté de pouvoir dégrader la cohésion du verre de conditionnement des déchets[42].
En revanche, il est établi qu'ils n'ont qu'une très faible mobilité dans les sols et l'environnement où ils se trouveraient dispersés[43]
Tableau de synthèse[modifier | modifier le code]
| Actinides par chaîne de désintégration | Période a |
Produits de fission par abondance de production | ||||||
|---|---|---|---|---|---|---|---|---|
| 4n | 4n+1 | 4n+2 | 4n+3 | |||||
| 2,25-3,5 % | 0,015-0,7 % | < 0,0065 % | ||||||
| 228Ra№ 0 | 4–6 | † | 155Euþ0 | |||||
| 244Cm 1 | 241Puƒ 1 | 250Cf 1 | 227Ac№ 1 | 10–29 | 90Sr 1 | 85Kr 1 | 113mCdþ 1 | |
| 232Uƒ 1 | 238Pu 1 | 243Cmƒ 1 | 29–97 | 137Cs 1 | 151Smþ 1 | 121mSn 1 | ||
| 249Cfƒ 2 | 242mAmƒ 2 | 141–351 |
Aucun produit de fission | |||||
| 241Am 2 | 251Cfƒ 2 | 430–900 | ||||||
| 226Ra№ 3 | 247Bk 3 | 1,3k–1,6k | ||||||
| 240Pu 3 | 229Th 3 | 246Cm 3 | 243Am 3 | 4,7k–7,4k | ||||
| 245Cmƒ 3 | 250Cm 3 | 8,3k–8,5k | ||||||
| 239Puƒ 4 | 24,11k | |||||||
| 230Th№ 4 | 231Pa№ 4 | 32k–76k | ||||||
| 236Npƒ 5 | 233Uƒ 5 | 234U№ 5 | 100k–250k | ‡ | 99Tc₡ 5 | 126Sn 5 | ||
| 248Cm 5 | 242Pu 5 | 280k–375k | 79Se₡ 5 | |||||
| 1,53M | 93Zr 6 | |||||||
| 237Np 6 | 2,1M–6,5M | 135Cs₡ 6 | 107Pd 6 | |||||
| 236U 7 | 247Cmƒ 7 | 15M–24M | 129₡ 7 | |||||
| 244Pu№ 7 | 80M |
Aucun atome au-dessus de 15,7 Ma | ||||||
| 232Th№ 9 | 238U№ 9 | 235Uƒ№ 9 | 0,703G–14G | |||||
|
Légende | ||||||||
Notes et références[modifier | modifier le code]
- (en) P. R. Fields, M. H. Studier, H. Diamond, J. F. Mech, M. G. Inghram, G. L. Pyle, C. M. Stevens, S. Fried, W. M. Manning, A. Ghiorso, S. G. Thompson, G. H. Higgins et G. T. Seaborg, « Transplutonium Elements in Thermonuclear Test Debris », Physical Review, vol. 102, no 1, , p. 180-182 (DOI 10.1103/PhysRev.102.180, Bibcode 1956PhRv..102..180F, lire en ligne)
- Les actinides mineurs représentent entre 2,7 % et 3,2 % en masse des produits de fission
- (en) CRC Handbook of Chemistry and Physics, section 1 : Basic Constants, Units, and Conversion Factors, sous-section : Electron Configuration of Neutral Atoms in the Ground State, 84e éd., en ligne, CRC Press, Boca Raton, Floride, 2003.
- (en) « Radioactive elements », sur International Union of Pure and Applied Chemistry (IUPAC) – Commission on Isotopic Abundances and Atomic Weights (CIAAW), (consulté le ).
- (en) Elena S. Craft, Aquel W. Abu-Qare, Meghan M. Flaherty, Melissa C. Garofolo, Heather L. Rincavage et Mohamed B. Abou-Donia, « Depleted and natural uranium: chemistry and toxicological effects », Journal of Toxicology and Environmental Health, Part B, vol. 7, no 4, , p. 297-317 (PMID 15205046, DOI 10.1080/10937400490452714, lire en ligne)
- (en) Rita Hindin, Doug Brugge et Bindu Panikkar, « Teratogenicity of depleted uranium aerosols: A review from an epidemiological perspective », Environmental Health, vol. 4, , p. 17 (PMID 16124873, PMCID 1242351, DOI 10.1186/1476-069X-4-17, lire en ligne)
- (en) Darryl P. Arfsten, Kenneth R. Still et Glenn D. Ritchie, « A review of the effects of uranium and depleted uranium exposure on reproduction and fetal development », Toxicology and Industrial Health, vol. 17, nos 5-10, , p. 180-191 (PMID 12539863, DOI 10.1191/0748233701th111oa, lire en ligne)
- (en) J. L. Domingo, J. L. Paternain, J. M. Llobet et J. Corbella, « The developmental toxicity of uranium in mice », Toxicology, vol. 55, nos 1-2, , p. 143-152 (PMID 2711400, DOI 10.1016/0300-483X(89)90181-9, lire en ligne)
- (en) Jay H. Lehr et Janet K. Lehr (2000), Standard handbook of environmental science, health, and technology, McGraw-Hill Professional, p. 2–38 (ISBN 0-07-038309-X).
- (en) « World Uranium Mining Production » (consulté le ).
- Fermi, E., « Possible Production of Elements of Atomic Number Higher than 92 », Nature, vol. 133, no 3372, , p. 898–899 (DOI 10.1038/133898a0, Bibcode 1934Natur.133..898F)
- Jagdish Mehra et Helmut Rechenberg, The historical development of quantum theory, Springer, (ISBN 978-0-387-95086-0, lire en ligne), p. 966–.
- Le nobélium et le lawrencium ont été découverts quasiment simultanément par les chercheurs américains et soviétiques.
- Martin Heinrich Klaproth, « Chemische Untersuchung des Uranits, einer neuentdeckten metallischen Substanz », Chemische Annalen, vol. 2, , p. 387–403 (lire en ligne)
- E.-M. Péligot, « Recherches sur l'uranium », Annales de chimie et de physique, vol. 5, no 5, , p. 5–47 (lire en ligne)
- Ingmar Grenthe, The Chemistry of the Actinide and Transactinide Elements, (DOI 10.1007/1-4020-3598-5_5), « Uranium ».
- Zimmerman, Ann., 213, 290 (1882) ; 216, 1 (1883) ; Ber. 15 (1882) 849
- Berzelius, J. J., « Untersuchung eines neues Minerals und einer darin erhalten zuvor unbekannten Erde (Investigation of a new mineral and of a previously unknown earth contained therein) », Annalen der Physik und Chemie, vol. 16, no 7, , p. 385–415 (DOI 10.1002/andp.18290920702, Bibcode 1829AnP....92..385B, lire en ligne) (modern citation: Annalen der Physik, vol. 92, n° 7, p. 385–415)
- Berzelius, J. J., « Undersökning af ett nytt mineral (Thorit), som innehåller en förut obekant jord (Investigation of a new mineral (thorite), as contained in a previously unknown earth) », Kungliga Svenska Vetenskaps Akademiens Handlingar (Transactions of the Royal Swedish Science Academy), , p. 1–30
- André-Louis Debierne, « Sur une nouvelle matière radio-active », Comptes rendus, vol. 129, , p. 593–595 (lire en ligne)
- André-Louis Debierne, « Sur une nouvelle matière radio-actif – l'actinium », Comptes rendus, vol. 130, 1900–1901, p. 906–908 (lire en ligne)
- H. W. Kirby, « The Discovery of Actinium », Isis, vol. 62, no 3, , p. 290–308 (DOI 10.1086/350760, JSTOR 229943)
- J. P. Adloff, « The centenary of a controversial discovery: actinium », Radiochim. Acta,, vol. 88, nos 3–4_2000, , p. 123–128 (DOI 10.1524/ract.2000.88.3-4.123)
- John Emsley, Nature's Building Blocks: An A-Z Guide to the Elements, Oxford, England, UK, Oxford University Press, (ISBN 0-19-850340-7, lire en ligne), « Protactinium », p. 347–349.
- Plutonium production, Federation of American Scientists.
- Reino W. Hakala, « Letters », Journal of Chemical Education, vol. 29, no 11, , p. 581 (DOI 10.1021/ed029p581.2, Bibcode 1952JChEd..29..581H)
- George B. Kauffman, « Victor Moritz Goldschmidt (1888–1947): A Tribute to the Founder of Modern Geochemistry on the Fiftieth Anniversary of His Death », The Chemical Educator, vol. 2, no 5, , p. 1–26 (DOI 10.1007/s00897970143a)
- Sections efficaces de l'233U, sur wwwndc.jaea.go.jp.
- 92-U-238 cross section table
- Sections efficaces du Pu 239
- 94-Pu-240 cross-section table
- 94-Pu-241 cross-section table
- 94-Pu-242 cross-section table
- Nucléide - Bibliothèque d'émission Laraweb : Liste des émissions pour l'américium 241
- 95-Am-241 cross-section table
- (en-US) A. Sasahara et al., « Neutron and Gamma Ray Source Evaluation of LWR High Burn-up UO2 and MOX Spent Fuels », Journal of Nuclear Science and Technology, vol. 41, no 4, , p. 448–456 (DOI 10.3327/jnst.41.448, lire en ligne) article/200410/000020041004A0333355.php Abstract
- 95-Am-241m cross-section table
- 95-Am-243 cross-section table
- tMLi : tonne de Métal Lourd initial.
- Voir l'article Chaîne de désintégration.
- Voir les articles Déchets radioactifs et Déchets radioactifs générés par la production d'électricité d'origine nucléaire en France.
- http://www.laradioactivite.com/fr/site/pages/Verres_R7T7.htm
- http://www.laradioactivite.com/fr/site/pages/RadiotoxiciteCU.htm
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Actinide » (voir la liste des auteurs).
Voir aussi[modifier | modifier le code]
Articles connexes[modifier | modifier le code]
Liens externes[modifier | modifier le code]
- « UICPA » (consulté le ) : Tableau périodique officiel du ; page de liens vers le tableau périodique
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | ||||||||||||||||
| 1 | H | He | |||||||||||||||||||||||||||||||
| 2 | Li | Be | B | C | N | O | F | Ne | |||||||||||||||||||||||||
| 3 | Na | Mg | Al | Si | P | S | Cl | Ar | |||||||||||||||||||||||||
| 4 | K | Ca | Sc | Ti | V | Cr | Mn | Fe | Co | Ni | Cu | Zn | Ga | Ge | As | Se | Br | Kr | |||||||||||||||
| 5 | Rb | Sr | Y | Zr | Nb | Mo | Tc | Ru | Rh | Pd | Ag | Cd | In | Sn | Sb | Te | I | Xe | |||||||||||||||
| 6 | Cs | Ba | La | Ce | Pr | Nd | Pm | Sm | Eu | Gd | Tb | Dy | Ho | Er | Tm | Yb | Lu | Hf | Ta | W | Re | Os | Ir | Pt | Au | Hg | Tl | Pb | Bi | Po | At | Rn | |
| 7 | Fr | Ra | Ac | Th | Pa | U | Np | Pu | Am | Cm | Bk | Cf | Es | Fm | Md | No | Lr | Rf | Db | Sg | Bh | Hs | Mt | Ds | Rg | Cn | Nh | Fl | Mc | Lv | Ts | Og | |
| 8 | 119 | 120 | * | ||||||||||||||||||||||||||||||
| * | 121 | 122 | 123 | 124 | 125 | 126 | 127 | 128 | 129 | 130 | 131 | 132 | 133 | 134 | 135 | 136 | 137 | 138 | 139 | 140 | 141 | 142 | |||||||||||
| Métaux alcalins | Métaux alcalino-terreux | Lanthanides | Métaux de transition | Métaux pauvres | Métalloïdes | Non-métaux | Halogènes | Gaz nobles | Éléments non classés |
| Actinides | |||||||||
| Superactinides |