
Organolithien
Un organolithien, ou simplement lithien, est un composé organométallique présentant une liaison carbone–lithium. Ce sont des réactifs importants en synthèse organique couramment utilisés pour transférer leur chaîne carbonée ou leur atome de lithium à travers une addition nucléophile ou une déprotonation[1]. On utilise les organolithiens dans l'industrie pour l'amorçage de réactions de polymérisation anionique permettant de produire de nombreux élastomères, ainsi qu'en synthèse asymétrique dans l'industrie pharmaceutique[2]. Le méthyllithium CH3Li, le n-butyllithium CH3CH2CH2CH2Li et le phényllithium C6H5Li sont des exemples d'organolithiens.

En raison de la grande différence d'électronégativité entre les atomes de carbone et de lithium, la liaison C–Li est fortement ionique. La nature polaire de cette liaison fait des organolithiens de bons nucléophiles ainsi que des bases fortes. De nombreux organolithiens sont disponibles sur le marché pour usage de laboratoire, distribués dans des solvants aprotiques. Ce sont des composés très réactifs, susceptibles d'être également pyrophoriques.

Préparation[modifier | modifier le code]
La plupart des alkyllithiens simples et les amidures de lithium les plus courants sont disponibles sur le marché dans divers solvants et à diverses concentrations. Ils peuvent également être préparés au laboratoire par différentes méthodes. Les sections suivantes en présentent quelques-unes.
Réaction avec le lithium métallique[modifier | modifier le code]
La réduction d'halogénoalcanes par du lithium métallique permet de produire des alkyllithiens et aryllithiens simples[3] :
La préparation industrielle des organolithiens à l'aide de cette méthode fait intervenir le chlorure d'alkyle avec du lithium métallique contenant de 0,5 à 2 % de sodium. La réaction est fortement exothermique. Le sodium amorce l'addition radicalaire et augmente la vitesse de réaction[4]. La réaction ci-dessous est un exemple de préparation d'un organolithien vinylique selon cette méthode[5]. Le lithium peut ici être utilisé sous forme de poudre fine avec certains catalyseurs comme le naphtalène ou le 4,4’-di-tert-butylbiphényle (DTBB).
-
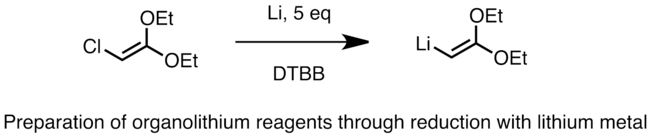 (en) Formation de 2,2-diéthoxyvinyllithium par réduction du chlorure.
(en) Formation de 2,2-diéthoxyvinyllithium par réduction du chlorure.

Outre les halogénures, il est également possible de produire des organolithiens à partir de sulfures, notamment pour obtenir des composés α-lithio[6].
Métallation[modifier | modifier le code]
Une seconde méthode permettant de préparer des organolithiens est la métallation. La position de la lithiation est contrôlée par l'acidité relative des atomes d'hydrogène sur la chaîne carbonée. C'est la méthode la plus courante pour produire des organolithiens car l'atome d'hydrogène terminal lié à un atome de carbone par une liaison sp est très acide, de sorte que cet atome de carbone est facilement déprotoné[3]. Pour les composés aromatiques, la position de la lithiation est également déterminée par la position des substituants[7]. Les plus directifs de ces substituants sont les groupes alcoxyle, amidure, sulfoxyde et sulfonyle. La métallation se produit souvent en position ortho de ces substituants. De même, la métallation se produit généralement en position ortho de l'hétéroatome dans les hétérocycles aromatiques[3],[7].
-
 (en) Exemples d'organolithiens produits par métallation.
(en) Exemples d'organolithiens produits par métallation.

Échange lithium-halogène[modifier | modifier le code]
Une troisième méthode permettant de préparer des organolithiens est par échange lithium-halogène. Le tert-butyllithium et le n-butyllithium sont les réactifs les plus couramment utilisés pour ce type de réactions. L'échange lithium-halogène est principalement utilisée pour convertir des iodures et bromures d'aryle et d'alkényle avec des atomes de carbone sp2 en organolithiens correspondants. La réaction est extrêmement rapide et se déroule généralement à des températures de −120 à −60 °C[8].
Transmétallation[modifier | modifier le code]
Une quatrième méthode de préparation des organolithiens est la transmétallation. C'est par exemple le cas pour préparer du vinyllithium CH2=CHLi.
Réaction de Shapiro[modifier | modifier le code]
Au cours de la réaction de Shapiro (en), deux équivalents de bases fortes d'alkyllithiens réagissent avec des composés p-tosylhydrazone (en) pour produire du vinyllithium ou, par trempe, l'alcène correspondant.
Structure et réactivité[modifier | modifier le code]
Bien que les alkyllithiens simples soient souvent représentés comme des monomères de formule générique R–Li, ils se présentent en réalité sous forme d'oligomères ou de polymères[9]. Le degré de polymérisation dépend des chaînes carbonées et de la présence d'autres ligands[10],[11]. Ces structures ont été élucidées à l'aide de diverses méthodes, notamment par spectroscopie RMN à 6Li, 7Li et 13C, ainsi que par cristallographie aux rayons X[1], et ont été confirmées par la chimie numérique[9].
Polarité de la liaison[modifier | modifier le code]

La liaison C–Li est fortement polaire en raison de la différence d'électronégativité entre les atomes de carbone et de lithium[12],[13],[14]. Certains organolithiens présentent cependant des propriétés telles que la solubilité dans les solvants apolaires qui compliquent la question[12]. La plupart des résultats suggèrent que la liaison C–Li est essentiellement ionique, cependant la question de savoir dans quelle mesure elle présente également un caractère covalent demeure discutée[13],[14]. Une estimation situe la composante ionique de la liaison C–Li des alkyllithiens entre 80 et 88 %[15], ce qui est bien davantage que celle estimée pour les réactifs de Grignard (organomagnésiens mixtes).
Dans les allylithiens, le cation de lithium est coordonné du côté de la liaison π de l'atome de carbone selon une configuration η3 et non à un carbanion portant une charge électrique localisée, ce qui fait que les allylithiens polymérisent généralement moins que les alkyllithiens[10],[16]. Dans les aryllithiens, le cation de lithium est coordonné à un carbanion localisé avec une liaison σ[10],[17].
Structure à l'état solide[modifier | modifier le code]

Comme de nombreuses autres espèces chimiques constituées de sous-unités polaires, les organolithiens forment des agrégats[11],[18]. La formation de ces agrégats est facilitée par les interactions électrostatiques, la coordination entre l'atome de lithium et les molécules de solvant environnantes ou les additifs polaires, et des effets stériques[11].
L'élément de base pour la formation de structures plus complexes est un centre carbanionique interagissant avec un triangle Li3 dans une configuration η- 3[9]. Dans les alkyllithiens simples, ces triangles s'agrègent pour former des structures tétraédriques ou octaédriques. Par exemple, le méthyllithium, l'éthyllithium et le tert-butyllithium sont tétramériques, de la forme [RLi]4. À l'état solide, le méthyllithium se présente sous la forme d'un tétramère de type cubane dans lequel quatre atomes de lithium forment un tétraèdre. Dans ce solide, chaque méthyle du tétramère peut avoir des interactions agostiques avec les cations de lithium d'autres tétramères adjacents[9],[11]. L'éthyllithium et le tert-butyllithium, quant à eux, ne présentent pas de telles interactions, et sont par conséquent solubles dans les solvants apolaires. D'autres alkyllithiens adoptent des structures hexamériques, comme le n-butyllithium, l'isopropyllithium et le cyclohexanyllithium[9].
-

-

-
 Polymère en échelle de phényllithium.
Polymère en échelle de phényllithium.




Des amidures de lithium comme le bis(triméthylsilyl)amidure de lithium (LiHMDS) et le diisopropylamidure de lithium (LDA) forment également des agrégats[19]. Les amidures de lithium adoptent des structures en échelle dans les solvants non coordinants et sont généralement sous forme de dimères dans les éthers. En présence de ligands fortement donneurs, il se forme des complexes trimériques ou tétramériques[20]. Par exemple, le LDA dans le THF forme avant tout des dimères[19]. La structure des amidures de lithium courants comme le LDA et le LiHMDS a été étudiée en détail par spectroscopie RMN[21].
Les silyllithiens, apparentés aux organolithiens mais avec une liaison silicium–lithium, forment un groupe de réactifs très utilisés pour former des complexes organométalliques et des dendrimères de polysilanes[11],[22]. Contrairement aux alkyllithiens, la plupart des silyllithiens tendent à former des structures monomériques coordonnées avec des molécules de solvant comme le THS, et peu d'entre eux se sont avérés former des agrégats oligomériques[11]. Cette différence peut s'expliquer par la méthode de préparation des silyllithiens, l'encombrement stérique provoqué par la présence de substituants alkyl volumineux sur l'atome de silicium, ainsi que par la nature moins polarisée des liaisons Si–Li. L'addition de ligands fortement donneurs comme le tétraméthyléthylènediamine (TMEDA) et la (-)-spartéine peut déplacer les molécules de solvant coordonnées aux silyllithiens[11].
Structure en solution[modifier | modifier le code]
Structure d'alkyllithiens dans divers solvants[10] Alkyllithien Solvant Structure Méthyllithium THF Tétramère Éther/HMPA Tétramère n-Butyllithium Pentane Hexamère Éther Tétramère THF Tétramère-dimère sec-Butyllithium Pentane Hexamère-tétramère tert-Butyllithium Pentane Tétramère THF Monomère Isopropyllithium Pentane Hexamère-tétramère Phényllithium Éther Tétramère-dimère Éther/HMPA Dimère
Les données structurelles établies à partir d'agrégats organolithiens dans des structures cristallines à l'état solide ne sont pas suffisantes pour savoir quelles structures ces molécules adoptent en solution compte tenu de la diversité d'environnements que celles-ci peuvent offrir[10]. Par ailleurs, la structure cristalline d'un organolithien peut être difficile à isoler. Par conséquent, étudier la structure en solution des organolithiens et des intermédiaires réactionnels contenant du lithium est particulièrement utile pour comprendre la réactivité de ces molécules[23]. La spectroscopie RMN s'est révélée être un outil puissant pour étudier les agrégats d'organolithiens en soluion. Pour les alkyllithiens, le couplage scalaire C–Li permet souvent de déterminer le nombre d'atomes de lithium interagissant avec un carbanion et si ces interactions sont statiques ou dynamiques[10]. Des signaux RMN distincts peuvent également distinguer la présence de plusieurs agrégats d'une même unité monomérique[24].
La structure des organolithiens est affectée par la présence de bases de Lewis telles que le tétrahydrofurane (THF), l'éther diéthylique (Et2O), le tétraméthyléthylènediamine (TMEDA) ou encore l'hexaméthylphosphoramide (HMPA)[9]. Le méthyllithium est un cas particulier, car sa solvatation dans les éthers ou la présence d'un additif polaire comme le HMPA ne désagrège pas sa structure tétramérique de l'état solide[11]. A contrario, le THF désagrège la structure hexamérique du n-butyllithium : Le tétramère est l'espèce principale, et la variation d'enthalpie libre ΔG pour l'interconversion entre tétramères et dimères vaut environ 46 kJ·mol-1[25]. Le TMEDA peut également chélater des cations de lithium du n-butyllithium et former des dimères solvatés comme [(TMEDA)LiBu-n)]2[9],[10]. On a pu montrer que phényllithium existe sous forme d'une tétramère distordu dans la forme solvatée cristallisée et comme mélange de dimères et de tétramères dans les solutions d'éther[10].
Structure et réactivité[modifier | modifier le code]
La réactivité et la sélectivité des organolithiens varie selon leur environnement chimique de la même façon que leur structure en solution[11],[26]. On a d'abord pensé que les organolithiens les moins agrégés en solution sont les plus réactifs[27], puis on a découvert des réactions faisant intervenir des dimères et d'autres oligomères[28], et on a pu montrer que les réactions faisant intervenir les dimères sont courantes pour les amidures de lithium comme le LDA[21]. Un ensemble d'études cinétiques de réactions en solution impliquant du LDA a montré les énolates faiblement agrégés n'induisent pas nécessairement une réactivité accrue[21].
Par ailleurs, certaines bases de Lewis accroissent la réactivité des organolithiens[29],[30]. Il n'est cependant pas toujours clair de savoir si ces adjuvants fonctionnent comme des ligands fortement chélatants et dans quelle mesure cet accroissement de réactivité résulte de modifications structurelles des agrégats[29],[30]. Par exemple, le TMEDA augmente la vitesse et le rendement de plusieurs réactions faisant intervenir des organolithiens[11]. Le TMEDA fonctionne comme un ligand donneur envers les alkyllithiens, réduisant leur degré d'agrégation[9], et augmentant leur caractère nucléophile[31]. Le TMEDA ne fonctionne cependant pas toujours comme un ligand donneur, particulièrement en présence d'anions d'oxygène et d'azote. Par exemple, il n'interagit que faiblement avec le LDA et le LiHMDS, et ce même en solution dans des hydrocarbures sans ligands donneurs susceptible d'entrer en compétition[32]. Lors de la lithiation d'une imine, alors que le THF se comporte comme un ligand fortement donneur avec le LiHMDS, le TMEDA se dissocie rapidement du LiHMDS, ce qui conduit à la formation de dimères de LiHMDS, qui sont la principale espèce à réagir. Par conséquent, dans le cas du LiHMDS, le TMEDA n'augmente pas la réactivité en réduisant le degré d'agrégation[33]. L'addition de HMPA aux amidures de lithium comme le LiHMDS et le LDA donne souvent un mélange de dimères et de monomères dans le THF. Cependant, le ratio [dimères]/[monomères] ne varie pas si l'on fait croître la concentration de HMPA, de sorte que l'augmentation de la réactivité ne provient pas de la désagrégation du réactif. Le mécanisme par lequel ces adjuvants accroissent la réactivité des organolithiens continue d'être investigué[26].
Basicité[modifier | modifier le code]
Le caractère anionique des lithiens en font des bases fortes, plus fortes que les magnésiens.
Quelques valeurs de pKa (constante d'acidité) :
| organolithiens | pKa |
|---|---|
| butyllithium | 40 |
| méthyllithium | 45 |
| tertbutyllithium | 50 |
Ce caractère fortement basique implique des précautions lors de la synthèse de ces composés (cf. paragraphe ci-après)
De plus, il offre une voie de synthèse au LDA (Diisopropylamidure de lithium), base forte très utile en chimie organique.
BuLi + (iPr)2NH → BuH + LDA
Synthèse[modifier | modifier le code]
La synthèse des organolithiens est semblable à celle des réactifs de Grignard. En voici quelques voies.
Insertion[modifier | modifier le code]
Les organolithiens les plus simples peuvent être obtenus par réaction entre le lithium et un dérivé halogéné. À cause de la grande réactivité du lithium et des organolithiens, il est indispensable d'utiliser des réactifs rigoureusement anhydres et de travailler en l'absence de dioxygène. Cette méthode s'apparente à la synthèse magnésienne, et nécessite deux équivalents de lithium.
Voici le bilan réactionnel :
R-X + 2 Li = R-Li + LiX
X représente un halogène, usuellement un chlore ou un brome
L'utilisation d'une cuve à ultrasons permet d'augmenter la vitesse de la réaction en provoquant un décapage du métal. Différents solvants peuvent être utilisés parmi lesquels les éthers et certains alcanes comme le pentane ou le cyclohexane.
Échange halogène-métal[modifier | modifier le code]
R-X + R'-Li → R-Li + R'-X
X represente un halogène
La force motrice de la réaction est l'obtention d'un composé plus stable.
La stabilité d'un organolithien est donnée par l'étude du carbanion associé. En effet, plus le carbanion est basique, moins il est stable.
Métallation[modifier | modifier le code]
La métallation est la substitution d'un atome d'hydrogène d'une molécule organique par un atome métallique.
Il est aisé d'effectuer des échanges hydrogène-métal car le butyllithium est un produit commercial.
Cette méthode est donc une bonne voie d'accès aux lithiens dérivés des alcynes, des aromatiques ou des composés vinyliques.
Réactions[modifier | modifier le code]
Leur réactivité s'apparente à celle des réactifs de Grignard. Cependant, la plus forte polarité de la liaison et l'absence de sels de magnésium permet d'éviter des réactions parasites qui ont lieu lors de l'utilisation des réactifs de Grignard.
Synthèse de cétones[modifier | modifier le code]
La réaction d'un réactif de Grignard sur un acide carboxylique est une réaction acido-basique. La réaction s'arrête car l'entité n'est alors plus assez réactive.
L'utilisation d'un organolithien permet de poursuivre la réaction, en obtenant ainsi une cétone.
Ouverture d'époxyde[modifier | modifier le code]
L'ouverture d'époxyde par un réactif de Grignard conduit souvent un mélange de plusieurs produits, dont des composés bromés et des composés réduits.
L'utilisation de composés lithiés permet d'éviter ces nombreuses réactions parasites.
Voici le mécanisme d'ouverture d'un époxyde. L'addition s'effectue sur le carbone le moins encombré.
Addition en 1,2 sur des cétones conjuguées[modifier | modifier le code]
L'addition d'un réactif de Grignard sur une alpha-énone produit un mélange de composé d'addition 1,4 et 1,2
L'utilisation d'un organolithien permet d'obtenir uniquement le composé d'addition 1,2.
L'oxygène porte le numéro 1 et puis l'indexation se poursuit de carbone en carbone. L'addition 1,2 signifie une addition de l'organométallique sur le carbone en alpha de l'oxygène.
Notes et références[modifier | modifier le code]
- (en) Jacob Zabicky, « Analytical Aspects of Organolithium Compounds », PATAI'S Chemistry of Functional Groups, (DOI 10.1002/9780470682531.pat0304, lire en ligne)
- (en) George Wu et Mingsheng Huang, « Organolithium Reagents in Pharmaceutical Asymmetric Processes », Chemical Reviews, vol. 106, no 7, , p. 2596-2616 (PMID 16836294, DOI 10.1021/cr040694k, lire en ligne)
- (en) Francis A. Carey, « Organometallic compounds of Group I and II metals », Advanced Organic Chemistry: Reaction and Synthesis, Pt. B, Springer, 2007. (ISBN 978-0-387-44899-2)
- (en) M. Schlosser, Organometallics in Organic Synthesis, Wiley, New York, 1994. (ISBN 0-471-93637-5)
- (en) Mohamed Si-Fodil, Humberto Ferreira, Jean Gralak et Lucette Duhamel, « Obtention of 2,2-(diethoxy) vinyllithium and 2-methyl-4-ethoxy butadienyl lithium by arene-catalysed lithiation of the corresponding chloro derivatives. Synthetic applications », Tetrahedron Letters, vol. 39, no 49, , p. 8975-8978 (DOI 10.1016/S0040-4039(98)02031-0, lire en ligne)
- (en) Theodore Cohen et Mahadevan Bhupathy, « Organoalkali compounds by radical anion induced reductive metalation of phenyl thioethers », Accounts of Chemical Research, vol. 22, no 4, , p. 152-161 (DOI 10.1021/ar00160a006, lire en ligne)
- (en) Victor Snieckus, « Directed ortho metalation. Tertiary amide and O-carbamate directors in synthetic strategies for polysubstituted aromatics », Chemical Reviews, vol. 90, no 6, , p. 879-933 (DOI 10.1021/cr00104a001, lire en ligne)
- (en) F. Leroux, M. Schlosser, E. Zohar et I. Marek, The Preparation of Organolithium Reagents and Intermediates, Wiley, New York, 2004. (ISBN 978-0-470-84339-0)
- (en) Thomas Stey et Dietmar Stalke, « Lead Structures in Lithium Organic Chemistry », PATAI'S Chemistry of Functional Groups, (DOI 10.1002/9780470682531.pat0298, lire en ligne)
- (en) Hans J. Reich, « Role of Organolithium Aggregates and Mixed Aggregates in Organolithium Mechanisms », Chemical Reviews, vol. 113, no 9, , p. 7130-7178 (PMID 23941648, DOI 10.1021/cr400187u, lire en ligne)
- (en) Viktoria H. Gessner, Christian Däschlein et Carsten Strohmann, « Structure Formation Principles and Reactivity of Organolithium Compounds », Chemistry A European Journal, vol. 15, no 14, , p. 3320-3334 (DOI 10.1002/chem.200900041, lire en ligne)
- (en) Eluvathingal D. Jemmis et G. Gopakumar, « Theoretical Studies in Organolithium Chemistry », PATAI'S Chemistry of Functional Groups, (DOI 10.1002/9780470682531.pat0297, lire en ligne)
- (en) Andrew Streitwieser, « Perspectives on Computational Organic Chemistry », The Journal of Organic Chemistry, vol. 74, no 12, , p. 4433-4446 (PMCID 2728082, DOI 10.1021/jo900497s, lire en ligne)
- (en) F. Matthias Bickelhaupt, Miquel Solà et Célia Fonseca Guerra, « Covalency in Highly Polar Bonds. Structure and Bonding of Methylalkalimetal Oligomers (CH3M)n (M = Li−Rb; n = 1, 4) », Journal of Chemical Theory and Computation, vol. 2, no 4, , p. 965-980 (PMID 26633056, DOI 10.1021/ct050333s, lire en ligne)
- (en) Erwin Weiss, « Structures of Organo Alkali Metal Complexes and Related Compounds », Angewandte Chemie International Edition, vol. 32, no 11, , p. 1501-1523 (DOI 10.1002/anie.199315013, lire en ligne)
- (en) Gideon Fraenkel et Fayang Qiu, « Observation of a Partially Delocalized Allylic Lithium and the Dynamics of Its 1,3 Lithium Sigmatropic Shift », Journal of the American Chemical Society, vol. 118, no 24, , p. 5828-5829 (DOI 10.1021/ja960440j, lire en ligne)
- (en) Gideon Fraenkel, Sheela Subramanian et Albert Chow, « The Carbon-Lithium Bond in Monomeric Aryllithiums: Dynamics of Exchange, Relaxation, and Rotation », Journal of the American Chemical Society, vol. 117, no 23, , p. 6300-6307 (DOI 10.1021/ja00128a020, lire en ligne)
- (en) Haakon Hope et Philip P. Power, « Isolation and crystal structures of the halide-free and halide-rich phenyllithium etherate complexes [(PhLi.Et2O)4] and [(PhLi.Et2O)3.LiBr] », Journal of the American Chemical Society, vol. 105, no 16, , p. 5320-5324 (DOI 10.1021/ja00354a022, lire en ligne)
- (en) Paul G. Williard et Joseph M. Salvino, « Synthesis, isolation, and structure of an LDA-THF complex », The Journal of Organic Chemistry, vol. 58, no 1, , p. 1-3 (DOI 10.1021/jo00053a001, lire en ligne)
- (en) Göran Hilmersson et Johan Granander, « Structure and Dynamics of Chiral Lithium Amides », PATAI'S Chemistry of Functional Groups, (DOI 10.1002/9780470682531.pat0342, lire en ligne)
- (en) David B. Collum, Anne J. McNeil et Antonio Ramirez, « Lithium Diisopropylamide: Solution Kinetics and Implications for Organic Synthesis », Angewandte Chemie International Edition, vol. 46, no 17, , p. 3002-3017 (PMID 17387670, DOI 10.1002/anie.200603038, lire en ligne)
- (en) Akira Sekiguchi, Vladimir Ya. Lee et Masato Nanjo, « Lithiosilanes and their application to the synthesis of polysilane dendrimers », Coordination Chemistry Reviews, vol. 210, no 1, , p. 11-45 (DOI 10.1016/S0010-8545(00)00315-5, lire en ligne)
- (en) Jocelyn M. Gruver, Lara R. Liou, Anne J. McNeil, Antonio Ramirez et David B. Collum, « Solution Structures of Lithium Enolates, Phenolates, Carboxylates, and Alkoxides in the Presence of N,N,N′,N′-Tetramethylethylenediamine: A Prevalence of Cyclic Dimers », The Journal of Organic Chemistry, vol. 73, no 19, , p. 7743-7747 (PMID 18781812, PMCID 2636848, DOI 10.1021/jo801532d, lire en ligne)
- (en) Hans J. Reich, D. Patrick Green, Marco A. Medina, Wayne S. Goldenberg, Birgir Ö. Gudmundsson, Robert R. Dykstra et Nancy H. Phillips, « Aggregation and Reactivity of Phenyllithium Solutions », Journal of the American Chemical Society, vol. 120, no 29, , p. 7201-7210 (DOI 10.1021/ja980684z, lire en ligne)
- (en) John F. McGarrity et Craig A. Ogle, « High-field proton NMR study of the aggregation and complexation of n-butyllithium in tetrahydrofuran », Journal of the American Chemical Society, vol. 107, no 7, , p. 1805-1810 (DOI 10.1021/ja00293a001, lire en ligne)
- (en) Hans J. Reich, « What’s Going on with These Lithium Reagents? », The Journal of Organic Chemistry, vol. 77, no 13, , p. 5471-5491 (PMID 22594379, DOI 10.1021/jo3005155, lire en ligne)
- (en) J. L. Wardell, « Chapter 2 », G. Wilinson, F. G. A. Stone et E. W. Abel, Comprehensive Organometallic Chemistry, vol. 1, 1re édition, Pergamon, New York, 1982. (ISBN 0080406084)
- (en) Carsten Strohmann et Viktoria H. Gessner, « Crystal Structures of n-BuLi Adducts with (R,R)-TMCDA and the Consequences for the Deprotonation of Benzene », Journal of the American Chemical Society, vol. 130, no 35, , p. 11719-11725 (PMID 18686951, DOI 10.1021/ja8017187, lire en ligne)
- (en) A. J. Chalk et T. J. Hoogeboom, « Ring metalation of toluene by butyllithium in the presence of N,N,N′,N′-tetramethylethylenediamine », Journal of Organometallic Chemistry, vol. 11, , p. 615-618 (DOI 10.1016/0022-328X(68)80091-9, lire en ligne)
- (en) Hans J. Reich et D. Patrick Green, « Spectroscopic and reactivity studies of lithium reagent-HMPA complexes », Journal of the American Chemical Society, vol. 111, no 23, , p. 8729-8731 (DOI 10.1021/ja00205a030, lire en ligne)
- (en) Michael A. Nichols et Paul G. Williard, « Solid-state structures of n-butyllithium-TMEDA, -THF, and -DME complexes », Journal of the American Chemical Society, vol. 115, no 4, , p. 1568-1572 (DOI 10.1021/ja00057a050, lire en ligne)
- (en) David B. Collum, « Is N,N,N',N'-tetramethylethylenediamine a good ligand for lithium? », Accounts of Chemical Research, vol. 25, no 10, , p. 448-454 (DOI 10.1021/ar00022a003, lire en ligne)
- (en) Max P. Bernstein et David B. Collum, « Solvent- and substrate-dependent rates of imine metalations by lithium diisopropylamide: understanding the mechanisms underlying krel », Journal of the American Chemical Society, vol. 115, no 18, , p. 8008-8018 (DOI 10.1021/ja00071a011, lire en ligne)
Voir aussi[modifier | modifier le code]
- Liaison carbone-carbone
- Liaisons chimiques du carbone avec d'autres éléments dans le tableau périodique :
| C-H | He | |||||||||||||||||
| C-Li | C-Be | C-B | C-C | C-N | C-O | C-F | Ne | |||||||||||
| C-Na | C-Mg | C-Al | C-Si | C-P | C-S | C-Cl | C-Ar | |||||||||||
| C-K | C-Ca | C-Sc | C-Ti | C-V | C-Cr | C-Mn | C-Fe | C-Co | C-Ni | C-Cu | C-Zn | C-Ga | C-Ge | C-As | C-Se | C-Br | C-Kr | |
| C-Rb | C-Sr | C-Y | C-Zr | C-Nb | C-Mo | C-Tc | C-Ru | C-Rh | C-Pd | C-Ag | C-Cd | C-In | C-Sn | C-Sb | C-Te | C-I | C-Xe | |
| C-Cs | C-Ba | * | C-Lu | C-Hf | C-Ta | C-W | C-Re | C-Os | C-Ir | C-Pt | C-Au | C-Hg | C-Tl | C-Pb | C-Bi | C-Po | C-At | Rn |
| Fr | C-Ra | * * |
Lr | Rf | Db | C-Sg | Bh | Hs | Mt | Ds | Rg | Cn | Nh | Fl | Mc | Lv | Ts | Og |
| ↓ | ||||||||||||||||||
| * | C-La | C-Ce | C-Pr | C-Nd | C-Pm | C-Sm | C-Eu | C-Gd | C-Tb | C-Dy | C-Ho | C-Er | C-Tm | C-Yb | ||||
| * * |
Ac | C-Th | C-Pa | C-U | C-Np | C-Pu | C-Am | C-Cm | C-Bk | C-Cf | C-Es | Fm | Md | No | ||||
| Liaison de base en chimie organique | Nombreuses utilisations en chimie |
| Recherche académique, peu d'usages courants | Liaison inconnue / non évaluée |