
Précurseur de parfum

Cet article est orphelin. Moins de trois articles lui sont liés ().
Vous pouvez aider en ajoutant des liens vers [[Précurseur de parfum]] dans les articles relatifs au sujet.
Les précurseurs de parfums sont des molécules peu ou non odorantes, de préférence non volatiles, qui libèrent de façon contrôlée des composés odorants volatiles à la suite d'une réaction chimique telle que la photolyse, l’oxydation, l’hydrolyse ; dans des conditions de la vie quotidienne, soit des conditions douces (pression atmosphérique, température ambiante ou s’en approchant)[1],[2],[3].
Généralités[modifier | modifier le code]
Les précurseurs de parfums sont utilisés dans le domaine de la parfumerie, que ce soit dans les parfums, mais aussi, voire surtout, dans des produits cosmétiques ou ménagers comme les lessives, les assouplissants… et permettent le relargage une grande variété de composés odorants: des alcools, des esters, des composés carbonylés (aldéhydes, cétones), etc. (voir figure 1 dans la section galerie) en fonction du précurseur choisi[4]. Ces molécules peuvent se distinguer en deux catégories :
- les « pro-fragrances », qui sont les précurseurs qui libèrent un seul composé odorant à la fois.
- les « pro-parfums », qui sont les précurseurs pouvant générer plusieurs composés odorants différents[2].
Le mécanisme de libération du composé odorant est généralement le suivant : le précurseur est formé du composé odorant (volatile) et du substrat, liés par une liaison covalente. À la suite d'une réaction chimique dans des conditions de réaction douces, la liaison covalente va être rompue, provoquant la libération du composé odorant et l’obtention du substrat (voir figure 2 dans la section galerie)[2]. Parmi les nombreuses réactions chimiques enclenchant la libération du composé odorant, la rupture d’une liaison covalente entre le substrat et la partie volatile odorante du précurseur par l’effet de la température est la première à avoir été étudiée[1].
Il s’agit par ailleurs d’un domaine de recherche assez jeune, les premiers brevets ayant été déposés dans les années 1980. Les premiers précurseurs étaient généralement assez simples chimiquement, avec, par exemple, l'utilisation d’esters ou d’acétals pour relarguer, respectivement, des alcools ou des aldéhydes parfumés[1]. Il existe une analogie dans le domaine pharmaceutique avec les prodrogues.
Intérêts[modifier | modifier le code]
En comparaison aux composés volatiles odorants traditionnels, dits « libres », les précurseurs de parfums présentent une durée de perception plus longue après application[2]. Dans le cas de certains précurseurs, la forme conjuguée présente une longévité plus importante et une plus grande résistance aux conditions de stockage par rapport à la forme libre du composé volatil. C’est le cas par exemple des aldéhydes odorants qui peuvent subir une oxydation ou une polymérisation selon les conditions de stockage. Le conditionnement sous forme de précurseur d’aldéhyde permet donc d’éviter ces dégradations et de rallonger la durée de vie du produit[5].
Types de relargage[modifier | modifier le code]
Hydrolyse non-enzymatique[modifier | modifier le code]
L'hydrolyse de précurseurs de parfum est une méthode de relargage contrôlé d'alcool odorants volatiles. Il en existe deux types, l'hydrolyse enzymatique et l'hydrolyse non-enzymatique. Les méthodes de relargages par hydrolyse non-enzymatique se distinguent des réactions d’hydrolyses enzymatiques par le large panel de volatils pouvant être concernés par une même réaction. En effet, les enzymes utilisées pour l’hydrolyse enzymatiques sont particulièrement sélective vis-à-vis des molécules dont elles catalysent la réaction[6].
Les méthodes d’hydrolyses non-enzymatiques utilisent la participation des groupes fonctionnels proches de la liaison hydrolysée, au sein de la même molécule. Cette catalyse par groupes fonctionnels proches est appelée catalyse intramoléculaire[6].
Par exemple, pour le relargage d’alcools odorants, le substrat et la substance volatile sont liés par une fonction ester portée par un cycle benzylique. En position ortho, un groupe nucléophile protoné, proche du groupe ester, catalyse l’hydrolyse de l’ester. En milieu neutre ou légérement basique, le précurseur est hydrolysé et relargue un alcool odorant[6]. Cette méthode a pu être appliquée pour des benzoates et également des maléates et des succinates, permettant le relargage d’alcools par changement du pH d’acide à légérement basique[7].
Le relargage par hydrolyse non-enzymatique est principalement utile lorsque le produit est conditionné en solution aqueuse et soumis à un changement de pH. Ces produits peuvent être soit conditionnés en solution acide (pour le gel douche et le shampooing), soit en solution basique (pour le savon). La réaction d’hydrolyse est déclenchée lors du rinçage, pendant lequel le pH du milieu devient neutre[4]. D’autres types de précurseurs conviennent à ce type de relargage : les esters inorganiques, les siloxanes, les acétals, les imines[4].
Hydrolyse enzymatique[modifier | modifier le code]
Le relargage des composés volatiles peut également se faire au contact des enzymes de la peau ou au contact des enzymes bactériennes présentes sur la peau. On distingue trois types de relargage enzymatique, toutes correspondant à un mécanisme d'hydrolyse[4],[8].
Relargage en présence de glycoside (précurseur)[modifier | modifier le code]
Cette méthode permet de relâcher des alcools à partir de glycosides, en particulier de monosaccharides et disaccharides grâce à l’action des enzymes glycosidases. L’intérêt de l’utilisation de ce précurseur ne se réduit pas uniquement à diminuer la volatilité du substrat dans l’air mais permet également un meilleur stockage et transport dû à une meilleure solubilité du précurseur dans l’eau par rapport au composé volatile hydrophobe. Ce qu’il faut savoir c’est que l’on trouve des glycosidases dans la peau, les plantes, les bactéries et les champignons[9].
Relargage par enzyme β-Lyase et aminoacylase[modifier | modifier le code]
Ces enzymes sont responsables des mauvaises odeurs dans le corps, en comprenant comment leur mécanisme fonctionne, on peut les utiliser pour séparer le volatile du substrat[9]. Le clivage du précurseur (voir figure 5 dans la section galerie) amène à la formation de plusieurs composés : l’ammoniac, le pyruvate et en fonction du précurseur utilisé, un alcool, un thiol ou un acide carboxylique[9].
Le clivage du carbamate (2) par une aminoacylase forme les composés suivants : un dioxyde de carbone, de la glutamine (acide aminé) et un alcool[9].
Relargage par hydrolase[modifier | modifier le code]
Les hydrolases sont présentes sur la peau ou dans les bactéries cutanées. Elles peuvent hydrolyser les triglycérides ce qui les rend utiles dans les produits d’entretien pour leur action détachante. Elles sont également utilisées pour déclencher le relargage par hydrolyse de certains précurseurs[10].
Elles servent à relarguer des alcools à partir d’esters carboxyliques, de carbonates, de sulfates, sulfites, phosphates. De même, pour relarguer des aldéhydes et cétones à partir d’énolates d’ester et des oximes à partir de carbonates d’oximes[10].
Photolyse[modifier | modifier le code]
La lumière du soleil est une source d'énergie naturelle des plus abondantes et importantes impliquées dans les processus biologiques comme la photosynthèse par exemple. La lumière possède des propriétés corpusculaires, cela permet aux photons d'interagir avec la matière.
Généralement, on applique du parfum sur des surfaces exposées à la lumière du jour. De ce fait, des précurseurs photosensibles peuvent être utilisés pour la libération contrôlée de composés volatils. Pour pouvoir améliorer l'efficacité de cette réaction de relargage dans des conditions douces, celle-ci doit pouvoir se faire avec la lumière naturelle. Il est également fréquent d’utiliser une solution polaire en présence d’oxygène en fonction de la réaction photochimique visée[11]. Lorsqu'on effectue une irradiation avec de la lumière UV : aux environs de 350–370 nm, l'arrachement radicalaire d’un atome d’hydrogène en position gamma dit “γ-hydrogène” forme un intermédiaire qui se nomme: 1,4-biradical (voir figure 7 dans la section galerie)[11]. Ce dernier va se décomposer pour donner les produits finaux de la réaction. Selon l'absence ou la présence d’oxygène dans le milieu réactionnel, un dégagement gazeux respectivement de CO ou de CO2 est observé[11]. En fonction de la structure du précurseur, et de la réaction photochimique, on obtient les aldéhydes et/ou les cétones correspondant à la suite d'une irradiation avec une lampe au xénon dans des conditions expérimentales ou à la lumière du soleil dans des conditions de la vie quotidienne[11]. La 2-oxo-2-phenylacetate et la 2-Cyclohexyl-2-oxoacetate (voir figure 8 dans la section galerie) ont été proposés comme étant des précurseurs appropriées pour les applications recherchées en parfumerie grâce à leur forme très similaire et avantageux offrant un relargage efficace[11]. Il existe une corrélation entre la quantité de composé odorant libérée et l’intensité de la lumière. Le citronellal est un bon exemple qui illustre ce phénomène de libération. En effet, la libération de cette molécule est davantage importante entre 12-13h, heure suivie d’une luminosité solaire maximale[12].
La photoisomérisation est un procédé photochimique entraînant l'isomérisation de la molécule. Autrement dit, l'arrivée d'un photon a pour effet de transformer l'isomère cis en isomère trans ou inversement, par une rotation autour de la double liaison. On retrouve dans le Tonkarose, le o-Hydroxy cinnamate de l'isomère E, suivie d’une chaîne carbonée. À la suite d’une exposition à la lumière solaire, le o-Hydroxy cinnamate s’isomérise donnant l'isomère Z, suivie d’une lactonisation intramoléculaire nous donnant la coumarine qui est odorante et d’un alcool (voir figure 6 dans la section galerie). Expérimentalement, les composés ont été testé en présence et en l’absence de la lumière solaire, seuls les échantillons placé sous la lumière solaire ont pu émettre une odeur[13].
Oxydation[modifier | modifier le code]
L’oxydation permet la libération de composés carbonylés (volatiles) tels que des aldéhydes ou des cétones parfumés par une rupture oxydante d’une liaison C=C au sein du précurseur de départ, notamment dans des conditions de réactions douces[14],[15].
Il existe deux voies principales différentes de libération de composé odorants impliquant une réaction d’oxydation : l’autoxydation et la photo-oxydation.
L’auto-oxydation implique l’oxygène atmosphérique O2, dans des conditions douces. Celui-ci réagit avec le précurseur de parfum organique lors de la réaction radicalaire en chaîne. Lors de l’étape (3), le radical capte un atome d’hydrogène sur un autre composé organique, produisant le produit primaire : un hydroperoxyde, ainsi qu’un nouveau radical libre alkyle qui va répéter l'étape (2), poursuivant ainsi la chaîne. L’hydroperoxyde formé peut alors subir différentes réactions en fonction de sa structure, pouvant libérer une grande variété de composés parfumés. En fonction du choix du composé organique de départ, tous les composés produits lors de la réaction peuvent être d’intérêt olfactif. Les composés les plus sensibles à l’auto-oxydation sont ceux pouvant céder facilement des hydrogènes pour pouvoir former des radicaux libres, tels que les composés benzyliques, allyliques et éthérés. En effet, les radicaux alkyles qui en résultent sont stabilisés par effet mésomère[15]. Le cas de l'éther divinylique illustre de nombreuses voies de fragmentation existantes suivant l’obtention du produit primaire : un couple d'isomères d'hydroperoxydes allyliques. Ici, un seul des deux isomères obtenus pour illustrer les différentes réactions de décomposition. La fragmentation peut se produire via des voies homolytiques ou hétérolytiques :
- Par fragmentation homolytique, il y a formation d’un radical alcoxyle (1) par rupture de la liaison O-O du peroxyde. Par β-scission sont obtenues un composé carbonylé ou un ester, ainsi qu’un radical alkyle, pouvant réagir avec l’oxygène pour former un radical peroxyle, et éventuellement former des composés carbonylés supplémentaires[15]. Le radical alcoxyle (1) peut également prélever un hydrogène, formant un hémiacétal (ci-dessous) qui va se décomposer en un aldéhyde parent (2), utilisé pour la synthèse de l’éther divinylique, et un aldéhyde parfumé[15].
- Par fragmentation hétérolytique, il y a formation d’un hémiacétal par réarrangement de Hock, se décomposant ensuite en deux composés carbonylés et en deux autres molécules. comme visible ci-dessous[15]. La fragmentation peut également résulter de l’addition de radicaux peroxyles à l’éther divinylique. Cette addition produit un radical libre alkyle, qui réagit avec O2 pour former un radical peroxyle. Ce dernier, dans un environnement où des hydrogène libres sont disponibles, peut former un hydroperoxyde[15]. On obtient des 1,2-diperoxydes pouvant se décomposer de façon homolytique en composés carbonylés ainsi qu’en formiates d’énols, dont l’hydrolyse permettrait de générer un aldéhyde parent (2) de l’éther divinylique[15]. Le choix de l’auto-oxydation permet de tirer profit de l’omniprésence du dioxygène dans l’atmosphère comme élément déclencheur d’une libération douce et lente[15],[16]. Ainsi, ces précurseurs peuvent être hydrolytiquement stables, contrairement à des précurseurs de parfum plus traditionnels, et donc seront plus facilement utilisables dans des produits de consommation à base aqueuse[15]. Néanmoins, cette même omniprésence pose problème car elle rend difficile le contrôle de l’auto-oxydation[16].
La photo-oxydation, généralement appelée photofragmentation de type II de Norrish, est particulièrement adaptée pour la libération contrôlée de composés carbonylés volatiles, mais nécessite que le précurseur soit sensible à la lumière en plus d’être sensible à l’oxygène, et que le solvant soit polaire[14]. Cette réaction se déroule en deux étapes, imagées ci-dessus : la photoréaction de type II de Norrish du précurseur de parfum, suivie de son oxydation. Les groupes de précurseur compatibles avec ce type de réactions sont les alkyles, les phényls, les cétones et les a-cétoesters. La photoréaction de type II de Norrish étant générale, elle permet de nombreuses variations structurelles du substrat, et donc l’obtention de nombreux composés carbonylés odorants[14],[16]. Il est également possible d’obtenir des aldéhydes et cétones par oxydation de β-aminoalcools par NaIO4 ou NaBiO3 non toxique[14],[16].
Rétro-1,4-Additions[modifier | modifier le code]
Les rétro-1,4-additions (ou de Michael) sont un type de réaction organique au cours de laquelle une molécule est clivée au niveau d'une liaison carbone-carbone, ce qui résulte en la formation de deux molécules plus petites. Plus précisément, ce type de réaction implique l'inversion d'une réaction d'addition 1,4, qui est un type courant de réaction d'addition en chimie organique[17].
La rétro-1,4-addition permet la libération de diverses cétones à partir de certains précurseurs, et est très utilisée pour libérer des cétones α,β-insaturées telles que les damascones et les ionones, représentant une voie de libération utile[17],[18].
Exemples :
- Précurseur thioether-(+ )-1, qui permet, selon le mécanisme présenté ci contre, d'obtenir du b-damascone[19].
- L'oxoester est un type de précurseur permettant, à la suite d'une retro-addition de Michael, de relarguer du a-damascone[19].
Galerie[modifier | modifier le code]
-
 Figure 1: Exemple de composés odorants.
Figure 1: Exemple de composés odorants. -
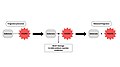 Figure 2: Mécanisme général de libération de composés odorants en utilisant un précurseur de parfum.
Figure 2: Mécanisme général de libération de composés odorants en utilisant un précurseur de parfum. -
 Figure 3: Schéma d'hydrolyse d'un benzoate précurseur de parfum.
Figure 3: Schéma d'hydrolyse d'un benzoate précurseur de parfum. -
 Figure 4: structure d'un monosaccharide.
Figure 4: structure d'un monosaccharide. -
 Figure 5 : Réaction enzymatique (β-Lyase).
Figure 5 : Réaction enzymatique (β-Lyase). -
 Figure 6: Réaction enzymatique (aminoacylase).
Figure 6: Réaction enzymatique (aminoacylase). -
 Figure 7: Formation de l'espèce 1,4-biradicale par irradiation UV.
Figure 7: Formation de l'espèce 1,4-biradicale par irradiation UV. -
 Figure 8: Structure de 2-oxo-2-phénylacétate et 2-Cyclohexyl-2-oxoacétate.
Figure 8: Structure de 2-oxo-2-phénylacétate et 2-Cyclohexyl-2-oxoacétate. -
 Figure 9: Libération du Coumarin odorant par photoisomérisation suivie d'une lactonisation intramoléculaire.
Figure 9: Libération du Coumarin odorant par photoisomérisation suivie d'une lactonisation intramoléculaire. -
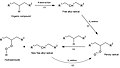 Figure 10: Mécanisme général d'autoxydation d'un précurseur de parfum.
Figure 10: Mécanisme général d'autoxydation d'un précurseur de parfum. -
 Figure 11: Réaction radicalaire en chaîne de l’auto-oxydation de l’éther divinylique.
Figure 11: Réaction radicalaire en chaîne de l’auto-oxydation de l’éther divinylique. -
 Figure 12 : Fragmentation homolytique avec β-scission.
Figure 12 : Fragmentation homolytique avec β-scission. -
 Figure 13 : Fragmentation homolytique avec décomposition d’un hémiacétal.
Figure 13 : Fragmentation homolytique avec décomposition d’un hémiacétal. -
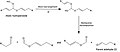 Figure 14 : Fragmentation hétérolytique.
Figure 14 : Fragmentation hétérolytique. -
 Figure 15: Addition de radicaux peroxyles aux liaisons C=C de l’éther divinylique dans un environnement avec des hydrogènes libres disponibles.
Figure 15: Addition de radicaux peroxyles aux liaisons C=C de l’éther divinylique dans un environnement avec des hydrogènes libres disponibles. -
 Figure 16: Photo-oxydation des a-cétoesters aux longueurs d’onde comprises entre 350 nm et 370 nm.
Figure 16: Photo-oxydation des a-cétoesters aux longueurs d’onde comprises entre 350 nm et 370 nm. -
![Figure 17 : Mécanisme de la rétro addition de type 1,4-Michael avec le thioéther (+-)-1[19].](//upload.wikimedia.org/wikipedia/commons/thumb/d/d0/Thioether_retro14.png/120px-Thioether_retro14.png) Figure 17 : Mécanisme de la rétro addition de type 1,4-Michael avec le thioéther (+-)-1[19].
Figure 17 : Mécanisme de la rétro addition de type 1,4-Michael avec le thioéther (+-)-1[19]. -
![Figure 18: Mécanisme de l'addition rétro de type 1,4-Michael avec oxoester[19].](//upload.wikimedia.org/wikipedia/commons/thumb/a/ae/Retro-1%2C4-Addition_oxoester.png/120px-Retro-1%2C4-Addition_oxoester.png) Figure 18: Mécanisme de l'addition rétro de type 1,4-Michael avec oxoester[19].
Figure 18: Mécanisme de l'addition rétro de type 1,4-Michael avec oxoester[19].







![Figure 17 : Mécanisme de la rétro addition de type 1,4-Michael avec le thioéther (+-)-1[19].](/wiki/Fichier:Thioether_retro14.png)
![Figure 18: Mécanisme de l'addition rétro de type 1,4-Michael avec oxoester[19].](/wiki/Fichier:Retro-1,4-Addition_oxoester.png)
Notes et références[modifier | modifier le code]
- (en) Keith D. Perring, « Perfumes », dans Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., (ISBN 978-0-471-23896-6, DOI 10.1002/0471238961.1605180619030818.a01.pub3, lire en ligne), p. 1–46
- (en) Andreas Herrmann, « Profragrances and Properfumes », The Chemistry and Biology of Volatiles, , p. 333 (ISBN 9780470777787)
- (en) F. Begnaud, J.-P. Larcinese, P. Fankhauser et U. Maddalena, « LogP measurement of a highly hydrophobic properfume: evaluation of extrapolation of RP-HPLC results and impact of chemical moieties on accuracy: LogP> 9: Impact of Chemical Moieties on extrapolation of HPLC measures », Flavour and Fragrance Journal, vol. 31, no 3, , p. 235–240 (DOI 10.1002/ffj.3309, lire en ligne, consulté le )
- (en) Andreas Herrmann, « Controlled Release of Volatiles under Mild Reaction Conditions: From Nature to Everyday Products », Angewandte Chemie International Edition, vol. 46, no 31, , p. 5836–5863 (DOI 10.1002/anie.200700264, lire en ligne, consulté le )
- (en) Alain Trachsel, Barbara Buchs et Andreas Herrmann, « Photolabile acetals as profragrances: the effect of structural modifications on the light-induced release of volatile aldehydes on cotton », Photochemical & Photobiological Sciences, vol. 15, no 9, , p. 1183–1203 (ISSN 1474-9092, DOI 10.1039/c6pp00206d, lire en ligne, consulté le ).
- Herrmann, Andreas., The Chemistry and Biology of Volatiles., Wiley, (ISBN 978-1-119-95698-3 et 1-119-95698-6, OCLC 927509384, lire en ligne), p. 340
- Andreas Herrmann, « Profragrance Chemistry as an Interdisciplinary Research Area and Key Technology for Fragrance Delivery », CHIMIA, vol. 71, nos 7-8, , p. 414 (ISSN 2673-2424 et 0009-4293, DOI 10.2533/chimia.2017.414, lire en ligne, consulté le )
- Herrmann, Andreas., The Chemistry and Biology of Volatiles., Wiley, (ISBN 978-1-119-95698-3 et 1-119-95698-6, OCLC 927509384, lire en ligne), p. 335
- (en) Andreas Herrmann, « Controlled Release of Volatiles under Mild Reaction Conditions: From Nature to Everyday Products », Angewandte Chemie International Edition, vol. 46, no 31, , p. 5845 (DOI 10.1002/anie.200700264, lire en ligne, consulté le )
- (en) Andreas Herrmann, « Controlled Release of Volatiles under Mild Reaction Conditions: From Nature to Everyday Products », Angewandte Chemie International Edition, vol. 46, no 31, , p. 5846 (DOI 10.1002/anie.200700264, lire en ligne, consulté le )
- (en) Andreas Herrmann, « Profragrance Chemistry as an Interdisciplinary Research Area and Key Technology for Fragrance Delivery », CHIMIA, vol. 71, nos 7-8, , p. 416 (ISSN 2673-2424 et 0009-4293, DOI 10.2533/chimia.2017.414, lire en ligne, consulté le ).
- Andreas Herrmann, « Profragrance Chemistry as an Interdisciplinary Research Area and Key Technology for Fragrance Delivery », CHIMIA, vol. 71, nos 7-8, , p. 417 (ISSN 2673-2424 et 0009-4293, DOI 10.2533/chimia.2017.414, lire en ligne, consulté le )
- (en) Andreas Herrmann, « Controlled Release of Volatiles under Mild Reaction Conditions: From Nature to Everyday Products », Angewandte Chemie International Edition, vol. 46, no 31, , p. 5844 (DOI 10.1002/anie.200700264, lire en ligne, consulté le )
- Herrmann, Andreas., The Chemistry and Biology of Volatiles., Wiley, (ISBN 978-1-119-95698-3 et 1-119-95698-6, OCLC 927509384, lire en ligne), p. 346-350
- (en) Brinda Indradas, Christopher Hansen, Michael Palmer et Gary B. Womack, « Autoxidation as a trigger for the slow release of volatile perfumery chemicals: Autoxidation for slow release of perfumery volatiles », Flavour and Fragrance Journal, vol. 29, no 5, , p. 313–323 (DOI 10.1002/ffj.3207, lire en ligne, consulté le )
- (en) Andreas Herrmann, « Controlled Release of Volatiles under Mild Reaction Conditions: From Nature to Everyday Products », Angewandte Chemie International Edition, vol. 46, no 31, , p. 5841 (DOI 10.1002/anie.200700264, lire en ligne, consulté le )
- (en) Herrmann Andreas, The Chemistry and Biology of Volatiles., Wiley, (ISBN 978-1-119-95698-3 et 1-119-95698-6, OCLC 927509384, lire en ligne), p. 354-356.
- (en) Umberto Maddalena, Alain Trachsel, Peter Fankhauser et Damien L. Berthier, « Thioether Profragrances: Parameters Influencing the Performance of Precursor-Based Fragrance Delivery in Functional Perfumery », Chemistry & Biodiversity, vol. 11, no 11, , p. 1700–1733 (DOI 10.1002/cbdv.201400023, lire en ligne, consulté le ).
- (en) Charles Fehr et José Galindo, « Aldols byMichael Addition: Application of theretro-Michael Addition to the Slow Release of Enones », Helvetica Chimica Acta, vol. 88, no 12, , p. 3128–3136 (ISSN 0018-019X et 1522-2675, DOI 10.1002/hlca.200590252, lire en ligne, consulté le ).