
Réaction d'insertion

Une réaction d'insertion est une réaction chimique où une entité chimique (une molécule ou un fragment moléculaire) s'interpose dans une liaison chimique, en général d'une deuxième entité chimique :
- A + B–C → B–A–C
ce terme ne se rapporte qu'au résultat de la réaction, et n'indique aucun mécanisme en particulier. On peut observer des réactions d'insertion en chimie organique, en chimie inorganique ou en chimie organométallique. Dans les cas d'une liaison métal-ligand d'un complexe, ces réactions sont de nature organométallique et impliquent une liaison entre un métal de transition et un carbone ou un hydrogène[1]. On réserve en général ce terme dans le cas où la coordinence et le nombre d'oxydation du métal restent inchangés[2]. Lorsque la réaction est réversible, on appelle le retrait de l'entité de la liaison métal-ligand « extrusion » ou « élimination ».
Il existe deux géométries d'insertion communes, 1,1 et 1,2 (voir image ci-dessus). De plus, la molécule insérée peut agir comme nucléophile ou comme électrophile par rapport au complexe métallique[2].
Chimie organique
[modifier | modifier le code]Les réactions d'homologation, comme l'homologation d'ester de Kowalski[3] sont des exemples simples de processus d'insertion en synthèse organique. Dans la réaction d'Arndt-Eistert[4],[5], une unité méthylène est inséré dans une liaison carboxyle-carbone d'un acide carboxylique pour former l'acide suivant de la série homologue. Organic Syntheses donne l'exemple de la (S)-phénylalanine (acide 2-amino-3-phénylpropanoïque) t-BOC protégée réagissant à la suite avec la triéthylamine, le chloroformate d'éthyle et le diazométhane pour produire une α-diazocétone, qui réagit ensuite avec le trifluoroacétate d'argent/triéthylamine en solution aqueuse pour générer la forme t-BOC protégée de l'acide (S)-3-amino-4-phénylbutanoïque[6] :
 .
.

Du point de vue mécanisme[7], l' α-diazocétone subit une réarrangement de Wolff[8],[9] pour former un cétène (groupe) via un réarrangement 1,2. En conséquence le groupe méthyle en α- du groupe carboxyle dans le produit est le groupe méthylène qui vient du réactif diazométhane. Il a été montré que le réarrangement 1,2 conservait la stéréochimie du centre chiral du produit formé : la forme t-BOC protégée de la (S)-phénylalanine donne le produit (S) avec un excès énantiomérique d'au moins 99 %[6].
Une transformation proche est la réaction de Nierenstein dans laquelle un groupe méthylène venant du diazométhane est inséré dans une liaison carbone-chlore d'un chlorure d'acyle pour former une cétone α-chlorométhylée[10],[11]. Un exemple, publié en 1924, illustre cette réaction sur un chlorure de benzoyle substitué[12] :
 .
.

De façon sans doute surprenante, une α-bromoacétophénone est le produit minoritaire de cette réaction quand elle et effectuée avec un bromure de benzoyle, un dioxane dimérique étant alors le produit majoritaire[13].

Les azotures organiques peuvent aussi fournir des exemples de réaction d'insertion en synthèse organique, et, comme dans les exemples ci-dessous, le procédé de transformation induit la formation de diazote gazeux. Lorsque l'azoture de tosyle réagit avec le norbornadiène, une réaction d'expansion de cycle se produit, dans laquelle un atome d'azote est inséré dans une liaison carbone-carbone en α du pont[14] :
 .
.

Le réarrangement de Beckmann[15],[16] est un autre exemple de réaction d'expansion de cycle dans laquelle un hétéroatome est inséré dans une liaison carbone-carbone. La plus importante application de cette réaction est la conversion de la cyclohexanone en son oxime, qui se réarrange ensuite en milieu acide pour former l' ε-caprolactame[17], produit de base dans la fabrication du Nylon 6. La production annuelle du caprolactame dépasse 2 milliards de kilogrammes[18].

Les carbènes peuvent subir des réactions d'insertion intermoléculaires et intramoléculaires. Des fractions de cyclopentène peuvent être générées à partir de cétones à chaine suffisamment longue par réaction avec le triméthylsilyldiazométhane, (CH3)3Si–CHN2 :
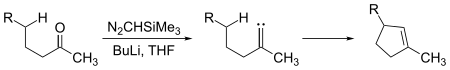 .
.

Ici, l'intermédiaire carbène s'insère dans une liaison carbone-hydrogène pour former la liaison carbone-carbone requise pour fermer le cycle de cyclopentène. Les insertions de carbène dans une liaison C-H peuvent être aussi intermoléculaires :
 .
.

Les carbénoïdes sont des intermédiaires réactionnels qui réagissent de façon similaire aux carbènes[19]. Un exemple est la réaction carbénoïde chloroalkyllithium préparé in situ à partir de sulfoxyde et de t-BuLi qui s'insère dans une liaison carbone-bore de l'ester boronique du pinacol[20] :
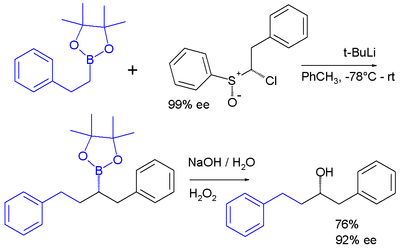 .
.

Chimie organométallique
[modifier | modifier le code]Beaucoup de réactions en chimie organométallique impliquent l'insertion d'un ligand (L) dans une liaison métal-hydrure ou métal-alkyle/aryle. En général, c'est l'hydrure, l'alkyle ou l'aryle qui migre sur L, qui est souvent un groupe CO, un alcène ou un alcyne.
Carbonylations
[modifier | modifier le code]L'insertion de monoxyde de carbone et d'alcènes dans une liaison métal-carbone est une réaction très largement exploitée avec des applications industrielles majeures[21],[22].

De telles réactions sont sujettes aux paramètres habituels qui affectent les autres réactions en chimie de coordination, mais les effets stériques sont particulièrement importants pour déterminer la stéréochimie et la régiochimie des réactions. Les réactions inverses, les « désinsertion » de CO et d'alcènes, sont d'une importance fondamentale dans beaucoup de cycles catalytiques également.
Les applications particulièrement courantes de l'insertion migratoire de groupes carbonyles sont l'hydroformylation (conversion d'alcènes, de dihydrogène et de monoxyde de carbone en aldéhydes) et la production carbonylative de l'acide acétique. Cette dernière peut s'effectuer par deux procédés industriels similaires. Le plus « traditionnel » est le procédé Monsanto à base de rhodium, mais il a été supplanté par le procédé Cativa à base d'iridium[23],[24]. En 2002, la production mondiale annuelle de l'acide acétique s'élève à 6 millions de tonnes, dont environ 60 % est produit par le procédé Cativa[23].
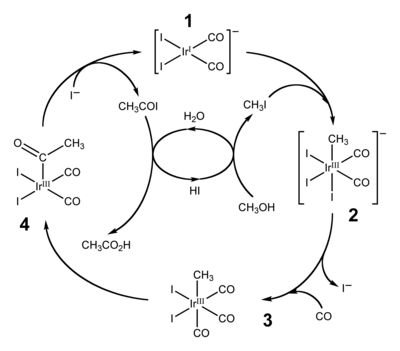
Cycle catalytique du procédé Cativa

Le cycle catalytique du procédé Cativa, montré ci-dessus, inclut à la fois des étapes d'insertion et de désinsertion. L'addition oxydante d'iodométhane avec (1) implique l'insertion formelle du centre d'iridium(I) dans la liaison carbone-iode, et les étapes (3) et (4) sont des exemples d'insertion migratoire du monoxyde de carbone dans la liaison iridium-carbone. Les espèces catalysatrices actives sont régénérées par l'élimination réductrice de l'iodure d'acétyle (4), une réaction de désinsertion[23].
Insertion d'alcènes
[modifier | modifier le code]L'insertion d'éthylène et de propylène dans des alkyles-titane est la pierre angulaire dans la catalyse de Ziegler-Natta, la voie de production commerciale du polyéthylène et du polypropylène. Cette technique implique principalement des catalyses hétérogènes, mais il est couramment admis que les principes et les observations sur des systèmes homogènes sont applicables aux versions à l'état solide. Parmi les techniques proches, on compte le « procédé SHOP (en) » qui produit des précurseurs de détergent[25]. Les alcènes peuvent être coordonnés au métal avant insertion. En dépendance avec la densité du ligand sur le métal, la dissociation de ligand peut être nécessaire pour fournir un site de coordination à l'alcène[26].

Autres réactions d'insertion en chimie de coordination
[modifier | modifier le code]Beaucoup d'oxydes électrophiles peuvent s'insérer dans une liaison métal-carbone ; on compte parmi eux le dioxyde de soufre, le dioxyde de carbone et les oxydes d'azote. Ces réactions ont des intérêts pratiques limités, mais présentent un intérêt historique. Ces oxydes se comportent comme des électrophiles avec les complexes alkyle-métal de transition et s'insèrent dans les liaisons entre les métaux et leurs ligands nucléophiles respectifs.
Notes et références
[modifier | modifier le code]- (en) Douglas, McDaniel et Alexander, Concepts and Models of Inorganic Chemistry 3rd Ed, John Wiley & Sons, Inc., , 1024 p. (ISBN 978-0-471-62978-8)
- (en) J.J. Alexander, The chemistry of the metal-carbon bond, vol. 2, Hartley and Patai,
- Ethyl 1-Naphthylacetate: Ester Homologation Via Ynolate Anions, Org. Synth. 71, coll. « vol. 9 », , 146 p., p. 426
- (de) F. Arndt et B. Eistert, « Ein Verfahren zur Überführung von Carbonsäuren in ihre höheren Homologen bzw. deren Derivate », Ber. Dtsch. Chem. Ges., vol. 68, no 1, , p. 200–208 (DOI 10.1002/cber.19350680142)
- (en) T. Ye et M. A. McKervey, « Organic Synthesis with α-Diazo Carbonyl Compounds », Chem. Rev., vol. 94, no 4, , p. 1091–1160 (DOI 10.1021/cr00028a010)
- (S)-3-(tert-Butyloxycarbonylamino)-4-phenylbutanoic acid, Org. Synth. 79, coll. « vol. 10 », , p. 194
- (en) C. Huggett, R. T. Arnold et T. I. Taylor, « The Mechanism of the Arndt-Eistert Reaction », J. Amer. Chem. Soc., vol. 64, no 12, , p. 3043 (DOI 10.1021/ja01264a505)
- (en) Meier, H. et Zeller, K.-P., « The Wolff Rearrangement of α-Diazo Carbonyl Compounds », Angew. Chem. Int. Ed., vol. 14, no 1, , p. 32–43 (DOI 10.1002/anie.197500321)
- (en) Kirmse, W., « 100 Years of the Wolff Rearrangement », Eur. J. Org. Chem., vol. 2002, no 14, , p. 2193–2256 (DOI 10.1002/1099-0690(200207)2002:14<2193::AID-EJOC2193>3.0.CO;2-D)
- (en) Clibbens, D. A. et Nierenstein, M., « The Action of Diazomethane on some Aromatic Acyl Chlorides », J. Chem. Soc., Trans., vol. 107, , p. 1491–1494 (DOI 10.1039/CT9150701491)
- (en) W. E. Bachmann et W. S. Struve, « The Arndt-Eistert Reaction », Org. React., vol. 1, , p. 38
- (en) Nierenstein, M., Wang, D. G. et Warr, J. C., « The Action of Diazomethane on some Aromatic Acyl Chlorides II. Synthesis of Fisetol », J. Amer. Chem. Soc., vol. 46, no 11, , p. 2551–2555 (DOI 10.1021/ja01676a028)
- (en) Lewis, H. H., Nierenstein, M. et Rich, E. M., « The Action of Diazomethane on some Aromatic Acyl Chlorides III. The Mechanism of the Reaction », J. Amer. Chem. Soc., vol. 47, no 6, , p. 1728–1732 (DOI 10.1021/ja01683a036)
- (en) D. D. Reed et S. C. Bergmeier, « A Facile Synthesis of a Polyhydroxylated 2-Azabicyclo[3.2.1]octane », J. Org. Chem., vol. 72, no 3, , p. 1024–1026 (PMID 17253828, DOI 10.1021/jo0619231)
- (de) E. Beckmann, « Zur Kenntniss der Isonitrosoverbindungen », Ber. Dtsch. Chem. Ges., vol. 19, , p. 988–993 (DOI 10.1002/cber.188601901222)
- (en) R. E. Gawley, « The Beckmann Reactions: Rearrangement, Elimination-Additions, Fragmentations, and Rearrangement-Cyclizations. », Org. React., vol. 35, , p. 14–24 (DOI 10.1002/0471264180.or035.01)
- ε-Benzoylaminocaproic acid, Org. Synth. 19, coll. « vol. 2 », , p. 76
- (en) J. Ritz, H. Fuchs, H. Kieczka et W. C. Moran, Ullmann's Encyclopedia of Industrial Chemistry, Weinheim, Wiley-VCH, (DOI 10.1002/14356007.a05_031), « Caprolactam »
- (en) J. McMurry, Organic Chemistry, Brooks/Cole, , 2e éd. (ISBN 0-534-07968-7)
- (en) P. R. Blakemore et M. S. Burge, « Iterative Stereospecific Reagent-Controlled Homologation of Pinacol Boronates by Enantioenriched-Chloroalkyllithium Reagents », J. Amer. Chem. Soc., vol. 129, no 11, , p. 3068–3069 (DOI 10.1021/ja068808s)
- Elschenbroich, C., Organometallics (2006) Wiley-VCH: Weinheim. (ISBN 978-3-527-29390-2)
- Hartwig, J.F., Organotransition Metal Chemistry, from Bonding to Catalysis ; University Science Books : New York, 2010. (ISBN 1-891389-53-X)
- (en) Jones, J. H., « The CativaTM Process for the Manufacture of Acetic Acid », Platinum Metals Rev., vol. 44, no 3, , p. 94–105 (lire en ligne)
- (en) Sunley, G. J. et Watson, D. J., « High Productivity Methanol Carbonylation Catalysis using Iridium - The CativaTM Process for the Manufacture of Acetic Acid », Catalysis Today, vol. 58, no 4, , p. 293–307 (DOI 10.1016/S0920-5861(00)00263-7)
- (en) Crabtree, R. H., The Organometallic Chemistry of the Transition Metals, Hoboken, N.J., John Wiley & Sons, , 505 p. (ISBN 978-0-470-25762-3, lire en ligne), p. 19–25
- (en) Y. V. Kissin, Alkene Polymerization Reactions with Transition Metal Catalysts, Amsterdam, Elsevier, , 207–290 p. (ISBN 978-0-444-53215-2, lire en ligne), « Synthesis, Chemical Composition, and Structure of Transition Metal Components and Cocatalysts in Catalyst Systems for Alkene Polymerization »
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Insertion reaction » (voir la liste des auteurs).